NGSからFASTQ形式のデータを取得したら、それをbowtieやHISAT2、STARなどのゲノムマッピングプログラムを用いて、リファレンスゲノム配列にマッピングして解析します。マッピング結果はマッピングファイル(SAM/BAM/CRAM形式)で取得されますが、ここではPythonを用いてマッピングファイルを操作する方法を説明していきます。
バイオインフォマティクス環境
OS
- Windows10 + WSL (WindowsユーザーのためのPythonを用いたゲノム解析環境)
Python
- Python 3.7.6
モジュール
- pysam 0.16.0.1
SAM形式/BAM形式/CRAM形式の構造
SAM形式/BAM形式やCRAM形式はアラインされたデータの表現形式として広く使われています。
SAM形式:基本となる非圧縮のテキストファイル
bowtieやHISAT2、STARなどのゲノムマッピングプログラムは出力がSAM形式となっているものが多くあります。SAM形式はアラインメントに関する情報が記載されたヘッダー部分と実際のアラインメントを格納しているアラインメント部分から構成されています。
なお、SAM形式の公式リファレンスは以下をご覧ください。
ヘッダー部分
ヘッダー部分は「@」から始まる以下の5つタグ(@HD, @SQ, @RG, @PG, @CO)から構成されています。それぞれのタグに含まれている代表的な項目も以下のようになります。
| タグ | 項目 | 説明 |
|---|---|---|
| @HD | ファイルのメタデータ | |
| VN | フォーマットバージョン | |
| SO | アラインメントのソートされた順番 「unknown(デフォルト)、unsorted, queryname, coordinate」のいずれかで示される | |
| @SQ | リファレンス配列の情報 | |
| SN | リファレンス配列名 | |
| LN | リファレンス配列の長さ | |
| AS | ゲノムアセンブリID | |
| M5 | 配列のMD5ファイルのチェックサム | |
| SP | 生物種 | |
| UR | 配列のURI | |
| @RG | リードグループ | |
| @PG | マッピングに用いられたプログラムの情報 | |
| ID | プログラムのID | |
| PN | プログラムの名前 | |
| CL | マッピングに用いられたコマンド | |
| DS | 説明 | |
| VN | プログラムのバージョン | |
| @CO | 一行コメント |
例えば以下で扱うサンプルファイルのヘッダー部分は以下のようになります。
@HD VN:1.0 SO:coordinate @SQ SN:1 LN:249250621 M5:1b22b98cdeb4a9304cb5d48026a85128 UR:ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/phase2_reference_assembly_sequence/hs37d5.fa.gz AS:NCBI37 SP:Human @SQ SN:2 LN:243199373 M5:a0d9851da00400dec1098a9255ac712e UR:ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/phase2_reference_assembly_sequence/hs37d5.fa.gz AS:NCBI37 SP:Human @SQ SN:3 LN:198022430 M5:fdfd811849cc2fadebc929bb925902e5 UR:ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/phase2_reference_assembly_sequence/hs37d5.fa.gz AS:NCBI37 SP:Human (省略) @SQ SN:GL000229.1 LN:19913 M5:d0f40ec87de311d8e715b52e4c7062e1 UR:ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/phase2_reference_assembly_sequence/hs37d5.fa.gz AS:NCBI37 SP:Human @SQ SN:GL000231.1 LN:27386 M5:ba8882ce3a1efa2080e5d29b956568a4 UR:ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/phase2_reference_assembly_sequence/hs37d5.fa.gz AS:NCBI37 SP:Human @SQ SN:GL000210.1 LN:27682 M5:851106a74238044126131ce2a8e5847c UR:ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/phase2_reference_assembly_sequence/hs37d5.fa.gz AS:NCBI37 SP:Human (省略) @RG ID:ERR034597 LB:HUMlsqXJXAAAPEI-8 SM:NA18942 PI:185 CN:BGI PL:ILLUMINA DS:SRP004076 @PG ID:bwa_index PN:bwa VN:0.5.9-r16 CL:bwa index -a bwtsw $reference_fasta (省略) @CO $known_indels_file(s) = ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/phase2_mapping_resources/ALL.wgs.indels_mills_devine_hg19_leftAligned_collapsed_double_hit.indels.sites.vcf.gz @CO $known_indels_file(s) .= ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/phase2_mapping_resources/ALL.wgs.low_coverage_vqsr.20101123.indels.sites.vcf.gz @CO $known_sites_file(s) = ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/phase2_mapping_resources/ALL.wgs.dbsnp.build135.snps.sites.vcf.gz
アラインメント部分
ヘッダー部分に続いてアラインメント部分となります。
1行につき一つのアラインメントを表し、各々の行はタブ区切りで左から順番に以下の基本11項目+αで構成されています。
| 番号 | 名前 | 説明 |
|---|---|---|
| 1 | QNAME | リード名 |
| 2 | FLAG | アラインメント結果 |
| 3 | RNAME | リファレンスゲノムの名前 |
| 4 | POS | リファレンスゲノム上におけるリードがマップされた開始位置 |
| 5 | MAPQ | マッピングクオリティスコア |
| 6 | CIGAR | CIGAR文字列(マッピング状況を表す文字列) |
| 7 | RNEXT | ペアエンドの場合、相手方のリード名 |
| 8 | PNEXT | ペアエンドの場合、相手方のマップされた開始位置 |
| 9 | TLEN | ペアエンドの場合、インサートの長さ |
| 10 | SEQ | マップした配列の塩基配列データ |
| 11 | QUAL | マップした配列のSequence Quality値 |
この11項目に関してはどのマッピングプログラムを用いても共通ですが、この後に続く項目はマッピングプログラム独自に定義された領域になります。
例えば以下で扱うサンプルファイルのアラインメント部分は以下のようになります。
ERR034597.57628075 163 20 60152 60 90M = 60213 121 TTTAAGAGATCCCATCACCCACATGAACGTTTGAATTGACAGGGGGAGCTGCCTGGAGAGTAGGCAGATGCAGCGCTCAAGCCTGTGCAG =??>BH@E@=FBA@@D=@AB@F@?F@=A;>BBHA??@G@F@IEFED9ECCGFBDHD?G?G?>FDE@G??GE?D*;@>6<@DC>BD>=C@# X0:i:1 X1:i:0 MD:Z:73A16 RG:Z:ERR034597 AM:i:37 NM:i:1 SM:i:37 MQ:i:60 XT:A:U BQ:Z:@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@ ERR034597.57628075 83 20 60213 60 29S61M = 60152 -121 GAATTGACAGGGGGAGCTGCCTGGAGAGTAGGCAGATGCAGAGCTCAAGCCTGTGCAGAGCCCAGGTTTTGTGAGTGGGACAGTTGCAGC ########################@A>AC>?ABC>BDE>DBEFFI@E@EHEFCEFFHEBGCBCAGAFDGABBD>HDGBADGCC@ X0:i:1 X1:i:0 XC:i:61 MD:Z:61 RG:Z:ERR034597 AM:i:37 NM:i:0 SM:i:37 MQ:i:60 XT:A:U BQ:Z:@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@ ERR034597.48086700 99 20 60270 60 80M10S = 60447 217 CAGCAAAACACAACCATAGGTGCCCATCCACCAAGGCAGGCTCTCCATCTTGCTCAGAGTGGCTCTAGCCCTTGCTGACTGCTGGGCAGG @=GEABBBGAGABGBA@?IE=GEAA@@FBAGAABIFEAHFEEFEEAA@GDBAEEGAGAH?GGFBEA;>2>AA>?BA3=A########### X0:i:1 X1:i:0 XC:i:80 MD:Z:80 RG:Z:ERR034597 AM:i:37 NM:i:0 SM:i:37 MQ:i:60 XT:A:U BQ:Z:@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@ ERR034597.48086700 147 20 60447 60 49S41M = 60270 -217 TTCTAATCTGGGCACCCCCCTCTCCACCCGACCCCGAGGCTCCACCCTGGCCACTCTTGTGTGCACACAGCACAGCCTCTACTGCTACAC ############################################AG>C@C@>A?@=*=<@6BABF1FDCDFAB?>F@DF=<E<< X0:i:1 X1:i:0 XC:i:41 MD:Z:41 RG:Z:ERR034597 AM:i:37 NM:i:0 SM:i:37 MQ:i:60 XT:A:U BQ:Z:@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@ ERR034597.30849717 99 20 60515 60 90M = 60811 385 CACTTTGTCCCCCCTGGCACAAATGGTGCTGGACCACGAGGGGCCAGAGAACAAAGCCTTGGGCATGGTCCCAACTCCCAAATGTTTGAA ==BBAAG>D;AAAACGDE@F?BB@HF?FFDHDAG?@A9=BD?DGACAAC@AAG:A?@6+8'.:<)52BBABECE7B@AA?F>AACA= X0:i:1 X1:i:0 MD:Z:64G25 RG:Z:ERR034597 AM:i:37 NM:i:1 SM:i:37 MQ:i:60 XT:A:U BQ:Z:@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@ ERR034597.37365692 99 20 60725 60 90M = 60950 314 TGTGCTTCTCTCTACAAGACTGGTGAGGGAAAGGTGTAACCTGTTTGTCAGCCACAACATCTTCCTAAGGGAGCCTTGTGTCCGGGAAAA @D>GFDAFDFCFD?GACIAGDHF>HAIFFACCIF>H?@CEBEH@BBH@GAIBCAGAAHA@GEBGCE@CIGFAHFCEBH?H?GC;FFABB@ X0:i:1 X1:i:0 MD:Z:90 RG:Z:ERR034597 AM:i:37 NM:i:0 SM:i:37 MQ:i:60 XT:A:U BQ:Z:@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@CGHH ERR034597.30849717 147 20 60811 60 90M = 60515 -385 AAAAACTGACAGACCAGTGATCTGGGTGCAGAAGGCTTGAGACAAAACTAGCTGGTTGGGCCAGCTATGGGGCAAATGCTGGAAAGAAAC =>>;?F:@?GCC5DG=>@E:<>?<=@:=A@A>@?53?@6C?=E5<==C9>:@?;@5>=@>0FCBH6?@??ADF>@>>?C=@=?>BD?<<= X0:i:1 X1:i:0 MD:Z:90 RG:Z:ERR034597 AM:i:37 NM:i:0 SM:i:37 MQ:i:60 XT:A:U BQ:Z:@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@ ERR034597.41549421 163 20 60821 60 90M = 61014 282 AGACCAGTGATCTGGGTGCAGAAGGCTTGAGACAAAACTAGCTGGTTGGGCCAGCTATGGGGCAAATGCTGGAAAGAAACCTGGTCAGGG =F?EA@H?G@@FDHFF=HF?IABIFFDBHAIAGACCCGE@FECFE>@G@EFCAIFC@?CFBEFABA?HFCHG?BBI?B@EBBDF?>?A?? X0:i:1 X1:i:0 MD:Z:90 RG:Z:ERR034597 AM:i:37 NM:i:0 SM:i:37 MQ:i:60 XT:A:U BQ:Z:@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@ ERR034597.42667768 99 20 60825 60 90M = 61023 287 CAGTGATCTGGGTGCAGAAGGCTTGAGACAAAACTAGCTGGTTGGGCCAGCTATGGGGCAAATGCTGGAAAGAAACCTGGTCAGGGAGCC @>GGFBGE=AED?DA?ADB>?F9@BD@AA>FCCHCAABDA::5?BD=>E@F?8:=9> X0:i:1 X1:i:0 MD:Z:90 RG:Z:ERR034597 AM:i:37 NM:i:0 SM:i:37 MQ:i:60 XT:A:U BQ:Z:@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@ ERR034597.52769015 99 20 60844 60 90M = 61068 313 GGCTTGAGACAAAACTAGCTGGTTGGGCCAGCTATGGGGCAAATGCTGGAAAGAAACCTGGTCAGGGAGCCTGAGCTGAGTGGTCCCCAC @CDCBH@H@GABBBGD?IFDHF?BHFEEB?GFE>@HEEFF@BB?HEEHFACCGACAHCDGG?EAGEAAG@CDGBGGDGAH?GF>ECBB@@ X0:i:1 X1:i:0 MD:Z:90 RG:Z:ERR034597 AM:i:37 NM:i:0 SM:i:37 MQ:i:60 XT:A:U BQ:Z:@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@ ERR034597.6094555 99 20 60878 60 90M = 61072 245 TGGGGCAAATGCTGGAAAGAAACCTGGTCAGGGAGCCTGAGCTGAGTGGTCCCCACAGTCATCTGCTTGGCAAGAAACCCTAGGTCGCAG =DADEE@BA?GEDGEABBHABBFBCFD?FAIFE@HFACG?HFDG@H>GE?FBAB@GAI?FA?FEFFD@FEEABG@AAEBBC?EE?F;F?G X0:i:1 X1:i:0 MD:Z:90 RG:Z:ERR034597 AM:i:37 NM:i:0 SM:i:37 MQ:i:60 XT:A:U BQ:Z:@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@I ERR034597.11395614 99 20 60895 60 90M = 60995 189 AGAAACCTGGTCAGGGAGCCTGAGCTGAGTGGTCCCCACAGTCATCTGCTTGGCAAGAAACCCTAGGTCGCAGGTGCTAGACCAGCTGCA @F?BBGBDGF?FAHFFAIFBDHAIFEGAI?HF?FBCBAGAH@FA@GEHFEBGFFACIA@AHCCE?HG?GGEC>GBGB;GFDGD@ X0:i:1 X1:i:0 MD:Z:90 RG:Z:ERR034597 AM:i:37 NM:i:0 SM:i:37 MQ:i:60 XT:A:U BQ:Z:@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@ ERR034597.5132361 1123 20 60895 60 90M = 60995 189 AGAAACCTGGTCAGGGAGCCTGAGCTGAGTGGTCCCCACAGTCATCTGCTTGGCAAGAAACCCTAGGTCGCAGGTGCTAGACCAGCTGCA @F?BBGBDHF>FAHFF@IFBCHAIFEHAI?HF?FBCBAGAI?FA@GEHFEBHFEACIACCFBCD@GFGED?GAGCAIEDHF@ X0:i:1 X1:i:0 MD:Z:90 RG:Z:ERR034597 AM:i:37 NM:i:0 SM:i:37 MQ:i:60 XT:A:U BQ:Z:@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@ ERR034597.37365692 147 20 60950 60 90M = 60725 -314 AGAAACCCTAGGTCGCAGGTGCTAGACCAGCTGCACACCCACAGCAACACTGCCATGCCCAGGATCCTCTGCCCTTGATCCTGAATCAAC B???7DDD=B?@BC?8C9FDBCDA>F>I@FFH?@FFFHABF@@FI@IAFFFHBAF@AFH@FA?@F@<= X0:i:1 X1:i:0 MD:Z:90 RG:Z:ERR034597 AM:i:37 NM:i:0 SM:i:37 MQ:i:60 XT:A:U BQ:Z:@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@
それぞれの行について、左からタブ区切りでQNAME、FLAG、RNAME、POS、…と対応していきます。
BAM形式/CRAM形式:SAM形式を圧縮したバイナリファイル
SAM形式は非圧縮のテキストファイルで、データのサイズが非常に大きくランダムアクセス性にも劣るので、通常はその圧縮版(バイナリ版)であるBAM形式が解析に用いられます。ゲノムマッピングプログラムから出力されたSAM形式はBAM形式に変換しておきましょう。なお、SAM形式とBAM形式は可逆圧縮で含まれている情報は全く同じであり、相互変換可能です。また、インデックスファイルも作成すると指定した染色体上でのアライン位置の検索など、ランダム検索を非常に高速に実行することができます。
また、BAMよりも圧縮効率のバイナリ圧縮形式のファイルとしてEBIで開発されたCRAM形式も用いられています。
サンプルで用いるBAMファイル
1000人ゲノムプロジェクトの公共データベースにBAM形式のファイルがありますので、今回はこのうち一つをサンプルとして用います。

ここでは、NA18942という日本人女性のデータを使いましょう。以下のページがNA18942のデータのダウンロードページなので、「1000 Genomes phase 3 release」を選択して、Data typesをAlignmentに設定します。
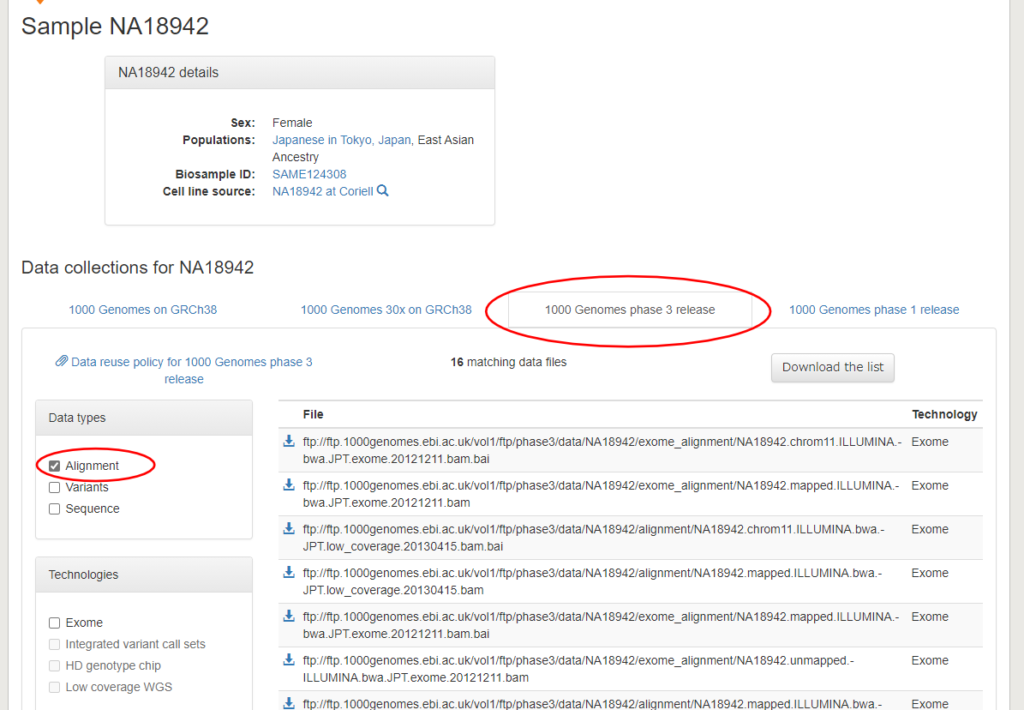
この一覧にあるファイルのうち、「***.bam」がマッピングファイルの本体で、「***.bai」がそのインデックスファイルとなっています。
すべてのゲノムデータにするとサイズが大きくなってしまうので、今回は20番染色体のデータに絞って解析を行ってみましょう。ファイル名の前にあるダウンロードマークを押して、以下のファイルをダウンロードします。
- ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/phase3/data/NA18942/exome_alignment/NA18942.chrom20.ILLUMINA.bwa.JPT.exome.20121211.bam
なお、ブラウザによってはアドレスバーに上記のFTPアドレスを直接指定してもダウンロード可能なものもあります。Linuxのcurlコマンドを使っても構いません。
Pythonでマッピングファイルを扱うモジュール
Pythonでマッピングファイルを扱うモジュールとしてはSAMtoolsのC言語APIのPythonラッパーであるpysamがあります。

以下では、pysamを用いたマッピングファイルの扱い方を説明していきます。
SAM形式/BAM形式から情報を取得する
SAM形式/BAM形式を読み込む
pysamではSAM形式/BAM形式のファイルをAlignmentFileオブジェクトとして読み込みます。AlignmentFileクラスのコンストラクタにパスを指定してインスタンス化するだけでSAM形式/BAM形式のファイルをPythonのオブジェクトとして読み込むことができます。なお、SAM形式であるかBAM形式であるかはファイル自体から自動で判別されるので、特に指定する必要はありません。
それでは、先ほどサンプルファイルとして取得したBAMファイルをPythonで読み込んでみましょう。なお、引数に指定するパスはファイルを保存した場所に合わせて適宜変更してください。
import pysam
with pysam.AlignmentFile('NA18942.chrom20.ILLUMINA.bwa.JPT.exome.20121211.bam') as bam_file:
# 処理
なお、上記の処理はリソースを確実に解放できるようにPythonのwith構文を用いていますが、以下のようにしても同じです。
import pysam
bam_file = pysam.AlignmentFile('BioTech-Lab/6371/NA18942.chrom20.ILLUMINA.bwa.JPT.exome.20121211.bam')
# 処理
bam_file.close()
ヘッダー部分を取得する
AlignmentFileオブジェクトのheader属性にヘッダー部分が辞書類似のデータ形式で格納されています。
まずは、ヘッダー部分をすべて取得してみましょう。
import pysam
with pysam.AlignmentFile('NA18942.chrom20.ILLUMINA.bwa.JPT.exome.20121211.bam') as bam_file:
print(bam_file.header)
@HD VN:1.0 SO:coordinate
@SQ SN:1 LN:249250621 M5:1b22b98cdeb4a9304cb5d48026a85128 UR:ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/phase2_reference_assembly_sequence/hs37d5.fa.gz AS:NCBI37 SP:Human
@SQ SN:2 LN:243199373 M5:a0d9851da00400dec1098a9255ac712e UR:ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/phase2_reference_assembly_sequence/hs37d5.fa.gz AS:NCBI37 SP:Human
@SQ SN:3 LN:198022430 M5:fdfd811849cc2fadebc929bb925902e5 UR:ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/phase2_reference_assembly_sequence/hs37d5.fa.gz AS:NCBI37 SP:Human
(省略)
@RG ID:ERR034597 LB:HUMlsqXJXAAAPEI-8 SM:NA18942 PI:185 CN:BGI PL:ILLUMINA DS:SRP004076
@PG ID:bwa_index PN:bwa VN:0.5.9-r16 CL:bwa index -a bwtsw $reference_fasta
@PG ID:bwa_aln_fastq PN:bwa PP:bwa_index VN:0.5.9-r16 CL:bwa aln -q 15 -f $sai_file $reference_fasta $fastq_file
@PG ID:bwa_sam PN:bwa PP:bwa_aln_fastq VN:0.5.9-r16 CL:bwa sampe -a 555 -r $rg_line -f $sam_file $reference_fasta $sai_file(s) $fastq_file(s)
(省略)
@CO $known_indels_file(s) = ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/phase2_mapping_resources/ALL.wgs.indels_mills_devine_hg19_leftAligned_collapsed_double_hit.indels.sites.vcf.gz
@CO $known_indels_file(s) .= ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/phase2_mapping_resources/ALL.wgs.low_coverage_vqsr.20101123.indels.sites.vcf.gz
@CO $known_sites_file(s) = ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/phase2_mapping_resources/ALL.wgs.dbsnp.build135.snps.sites.vcf.gz
これは辞書に類似したデータ形式となっており、例えば@HDタグの情報を取得する場合は、
import pysam
with pysam.AlignmentFile('NA18942.chrom20.ILLUMINA.bwa.JPT.exome.20121211.bam') as bam_file:
print(bam_file.header['HD'])
{'VN': '1.0', 'SO': 'coordinate'}
となります。さらに、そのタグの項目名まで指定する場合は次のようになります。
import pysam
with pysam.AlignmentFile('NA18942.chrom20.ILLUMINA.bwa.JPT.exome.20121211.bam') as bam_file:
print(bam_file.header['HD']['VN'])
1.0
なお、@SQタグや@PGタグのように同じタグ名のものが複数ある場合は、それぞれのタグがリストで格納されているので、次のように抽出したいタグの番号を数字で0から始まる指定します。
import pysam
with pysam.AlignmentFile('NA18942.chrom20.ILLUMINA.bwa.JPT.exome.20121211.bam') as bam_file:
print(bam_file.header['SQ'][1])
{'SN': '2', 'LN': 243199373, 'M5': 'a0d9851da00400dec1098a9255ac712e', 'UR': 'ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/phase2_reference_assembly_sequence/hs37d5.fa.gz AS:NCBI37 SP:Human'}
ただし、この出力を見ると@SQタグのURとAS、SPが正しく分離できていません。元ファイルのヘッダー情報が正しくタブ区切りで構成されていないのが原因のようです。
アラインメント部分を取得する
AlignmentFileオブジェクトにおけるアラインメント部分は、AlignedSegmentオブジェクトをアイテムとするイテレータとして格納されています。Pythonの組み込み関数のnext関数を用いて、AlignedSegmentオブジェクトを順番に取り出すことが可能です。
まずは最初のアラインメントを出力してみましょう。
import pysam
with pysam.AlignmentFile('NA18942.chrom20.ILLUMINA.bwa.JPT.exome.20121211.bam') as bam_file:
print(next(bam_file))
ERR034597.57628075 163 19 60151 60 90M 19 60212 90 TTTAAGAGATCCCATCACCCACATGAACGTTTGAATTGACAGGGGGAGCTGCCTGGAGAGTAGGCAGATGCAGCGCTCAAGCCTGTGCAG array('B', [28, 30, 30, 29, 33, 39, 31, 36, 31, 28, 37, 33, 32, 31, 31, 35, 28, 31, 32, 33, 31, 37, 31, 30, 37, 31, 28, 32, 26, 29, 33, 33, 39, 32, 30, 30, 31, 38, 31, 37, 31, 40, 36, 37, 36, 35, 24, 36, 34, 34, 38, 37, 33, 35, 39, 35, 30, 38, 30, 38, 30, 29, 37, 35, 36, 31, 38, 30, 30, 38, 36, 30, 35, 9, 26, 31, 29, 21, 27, 31, 35, 34, 29, 33, 35, 29, 28, 34, 31, 2]) [('X0', 1), ('X1', 0), ('MD', '73A16'), ('RG', 'ERR034597'), ('AM', 37), ('NM', 1), ('SM', 37), ('MQ', 60), ('XT', 'U'), ('BQ', '@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@')]
このアラインメントからそれぞれの情報を取り出してみましょう。これはAlignedSegmentオブジェクトなので、その属性で指定します。
import pysam
with pysam.AlignmentFile('NA18942.chrom20.ILLUMINA.bwa.JPT.exome.20121211.bam') as bam_file:
read = next(bam_file)
print('リード名:' + read.query_name)
print('リファレンスゲノム名:' + read.reference_name)
print('リファレンスゲノム上のマッピング開始位置:' + str(read.reference_start))
print('CIGAR文字列:' + read.cigarstring)
リード名:ERR034597.57628075 リファレンスゲノム名:20 リファレンスゲノム上のマッピング開始位置:60151 CIGAR文字列:90M
なお、SAM形式の元ファイルではリファレンスゲノム上のマッピング開始位置は60152となっていますが、これはSAM形式では1から始まる整数で順番に値が振られているのに対して、Python上では0から始まる整数で順番に値が振られていることが理由です。
SAM形式/BAM形式を変換する
SAM形式/BAM形式を相互に変換する
bowtieなどのゲノムマッピングプログラムでは出力されたファイルは非圧縮のテキスト形式であるSAM形式で得られることが多いので、これを解析・保存に適したBAM形式に変換しましょう。
SAM形式のファイル(sample.sam)をBAM形式(sample.bam)に変換するコードは以下のようになります。
import pysam
# sample.samを開きます。
with pysam.AlignmentFile('sample.sam') as sam_file:
# sample.samのヘッダー部分をコピーして、バイナリモードでsample.bamというファイルを作成します。
with pysam.AlignmentFile('sample.bam', mode='wb', template = sam_file) as bam_file:
# sample.samのリードを1つ1つsample.bamに追加していきます。
for read in sam_file:
bam_file.write(read)
また同様に、BAM形式のファイル(sample.bam)をSAM形式(sample.sam)に変換するコードは以下のようになります。
import pysam
# sample.bamを開きます。
with pysam.AlignmentFile('sample.bam') as bam_file:
# sample.bamのヘッダー部分をコピーして、テキストモードでsample.samというファイルを作成します。
with pysam.AlignmentFile('sample.sam', mode='w', template = bam_file) as sam_file:
# sample.bamのリードを1つ1つsample.samに追加していきます。
for read in bam_file:
sam_file.write(read)
なお、BAM形式/SAM形式のファイルが大きい場合は、処理に時間がかかることがありますのでご注意ください。
BAMファイルのソートする
pysamではSAMtools C-APIを経由して、SAMtoolsのコマンドを実行することもできます。詳しくは以下の公式ドキュメントをご覧ください。
これを用いて、BAMファイルのソートを行ってみましょう。BAMファイル(sample.bam)をソートしてsample_sorted.bamとして出力するSAMtoolsのコマンドは、
samtools sort sample.bam -o sample_sorted.bam
となります。pysamではpysam.sort関数という関数の引数に、samtools sortコマンドの引数をすべて指定していきます。
import pysam
pysam.sort('sample.bam', '-o','sample_sorted.bam')
samtools sortコマンド以外のコマンドも同様にpysam上から実行することが可能です。
BAMファイルのインデックスを作成する
BAMファイルのインデックス作成にはsamtools indexコマンドを用いますが、これも先ほどと同様にpysam上から行ってみましょう。
BAMファイル(sample_sorted.bam)のインデックスを作成する場合は以下のようにします。
import pysam
pysam.index('sample_sorted.bam')
これで、インデックスファイル(sample_sorted.bam.bai)が作成されました。








コメント