バリアントコールファイル(VCF形式)は一塩基多型(SNP)、挿入欠失(InDel)、コピー数多型(CNV)などのゲノム配列の変異情報を保存するためのデータ形式です。
ここではVCF形式の基本的な事項と、Pythonを用いた基本的な操作方法を説明してきます。
バイオインフォマティクス環境
OS
- Windows10 + WSL (WindowsユーザーのためのPythonを用いたゲノム解析環境)
Python
- Python 3.7.6
モジュール
- PyVCF 0.6.8
VCF形式の基本構造
VCF形式は次の3つの部分から構成されます。
- アノテーション領域:行頭が「##」で始まる行
- ヘッダー行:行頭が「#」で始まる行
- データ領域:ヘッダーに続く行
アノテーション領域にはそのVCFファイルのアノテーション情報が記録されています。ヘッダー行でデータ領域に記録されているデータについての説明がなされ、それに続くデータ領域の1つの行が1つの変異を示しています。
VCF形式のファイルからアノテーション領域を除いたヘッダー行とデータ領域のサンプルは以下のようになります。
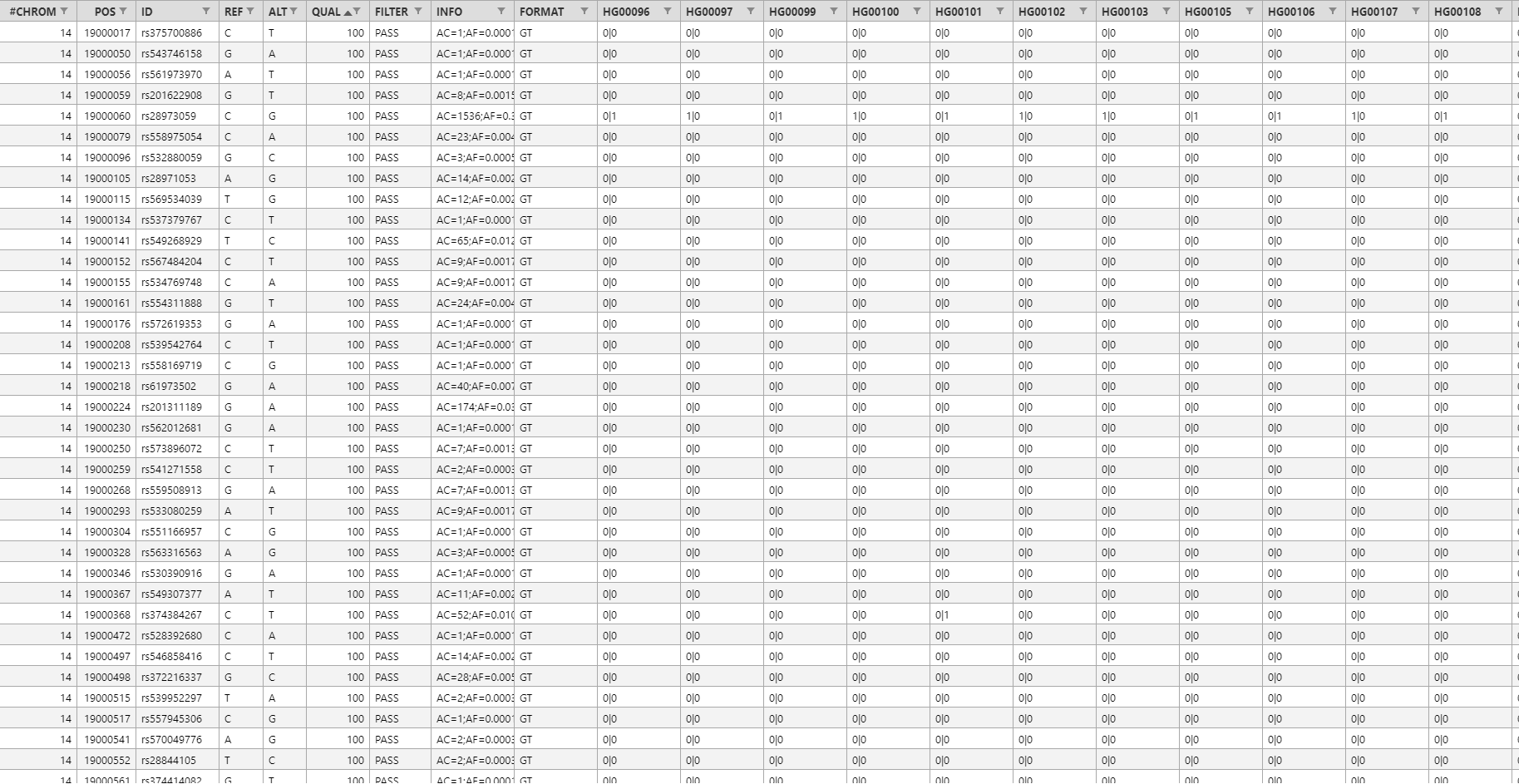
なお、実際のVCF形式のファイルはタブ区切りのテキスト形式ですが、この例では表形式で表示しています。
ヘッダー行に含まれる情報は左から順番に以下のようになります。
- CHROM:染色体番号
- POS:変異のある塩基の位置
- ID:変異のID (dbSNPに登録されている変異なら、そのアクセッション番号が記載される)
- REF:変異部位のリファレンスゲノム上での塩基配列
- ALT:変異部位のシークエンスデータ上での塩基配列
- QUAL:変異部位のマッピングクオリティ値(検出ソフトによって算出方法が異なる)
- FILTER:VCFファイルを作成する際に使用したフィルタリング条件を満たしているか
- INFO:付加情報(key=dataの形式でセミコロン区切りで複数指定可能、詳細は以下参照)
- FORMAT:10列目以降のデータのフォーマット(詳細は以下参照)
以上の1~9列目まではすべてのVCF形式のファイルで共通の項目ですが、10列目以降は各サンプルについての情報が記載されているのでVCFファイルごとに項目が異なります。先ほどの例では、FORMAT列で10列目以降のデータのフォーマットが定義されていて、そのフォーマットに従ってHG00096、HG00097、… という各サンプルにおける変異の情報が記載されています。VCF形式は複数のサンプルの変異情報をまとめて表示するためのものなので、例えば1000個のサンプルがある場合は10~1009列目が各サンプルについての情報となります。
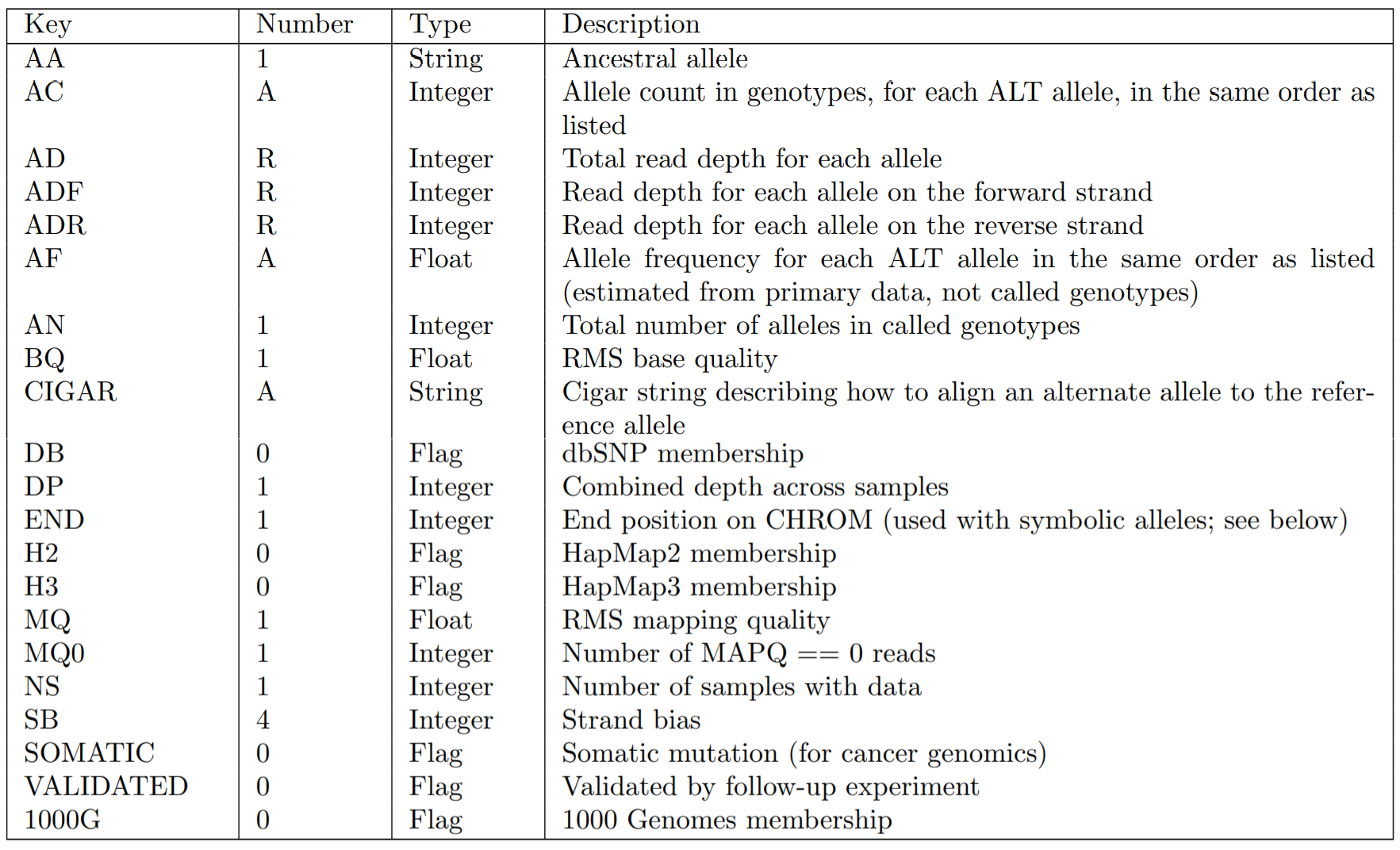
また、INFOで記載できる情報で定義済みのKeyは以下のようなものがあります。(これ以外でも独自に定義可能です)
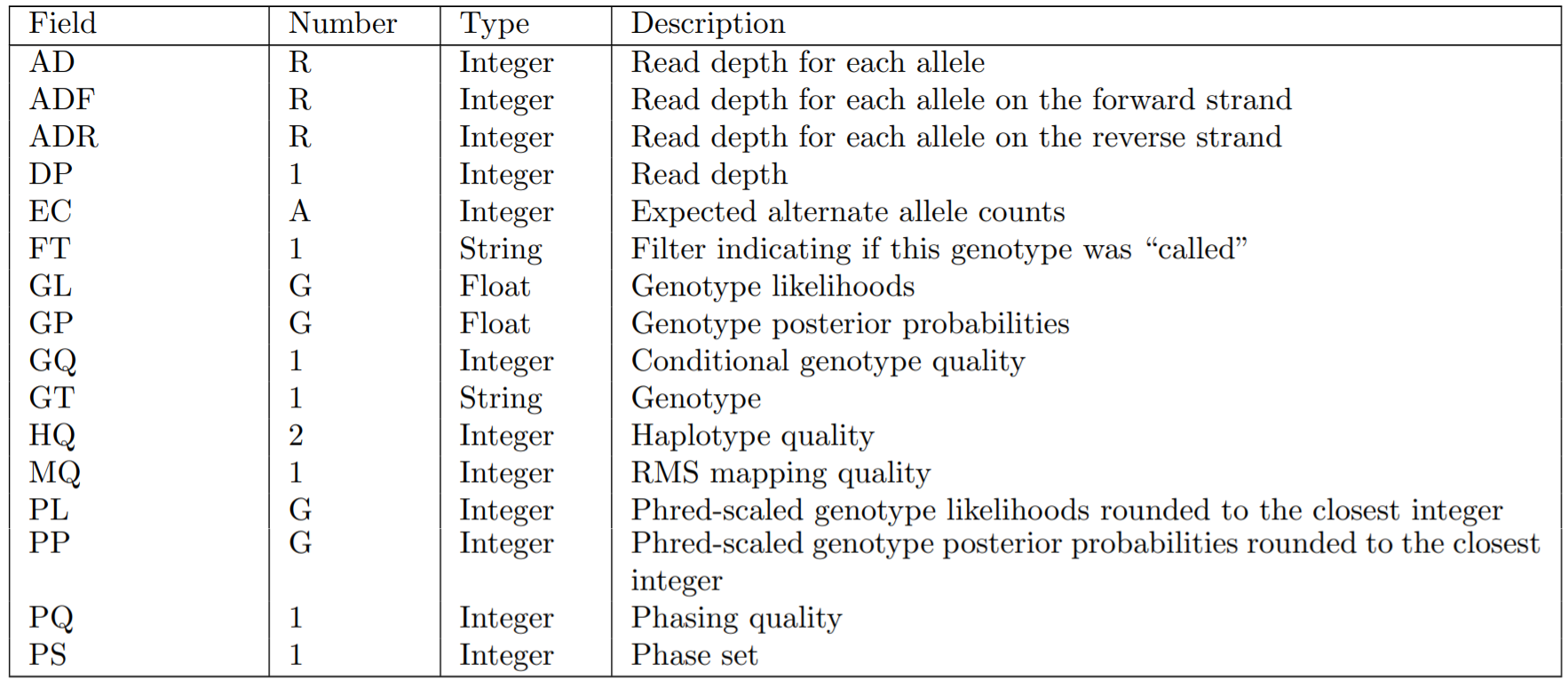
FORMATで指定できる情報は以下の通りです。
VCF形式についてのドキュメントは以下をご覧ください。
The Variant Call Format Specification
なお、VCF形式の詳細については以下のサイトに大変わかりやすく紹介されていましたので、ご紹介させていただきます。
サンプルで用いるVCFファイル
1000人ゲノムプロジェクトの公共データベースにVCF形式のファイルがありますので、今回はこのうち一つをサンプルとして用います。

ここでは、1000 Genomes phase 3 releaseのデータセットを使いましょう。VCF形式ではすべてのサンプルにおける変異情報がまとめられているので、どのサンプルのダウンロードページからも同じデータが得られます。例えば、日本人女性のサンプル(NA18942)のデータのダウンロードページは以下で、ここで「1000 Genomes phase 3 release」を選択して、Data typesをVariantsに設定します。
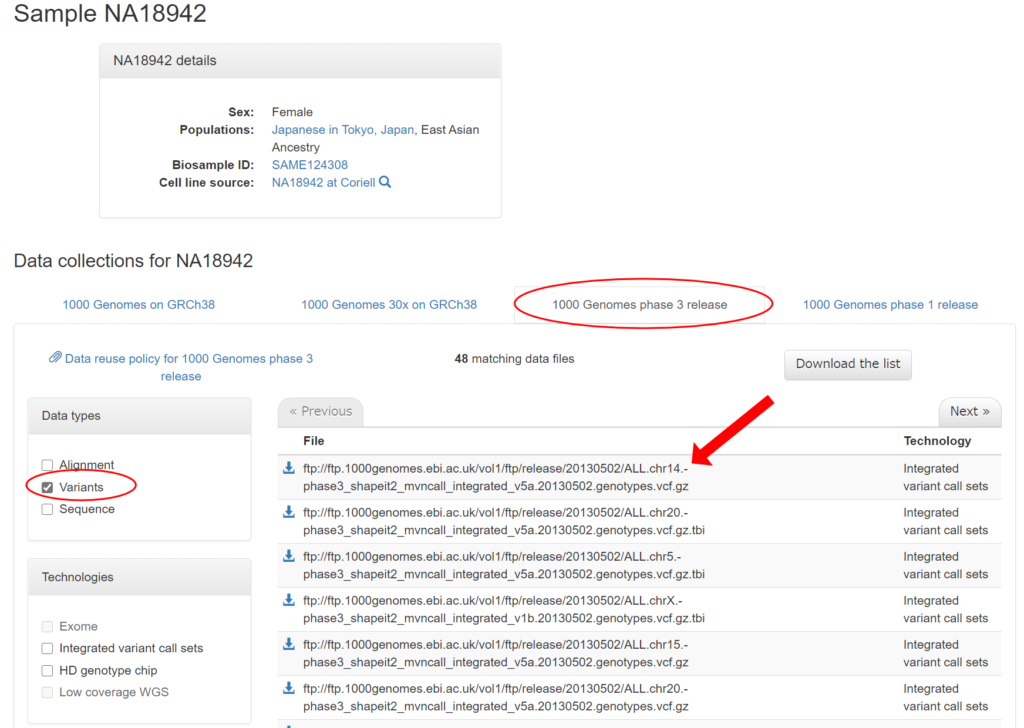
今回は一番上にある14番染色体のデータを使うので、ファイル名の前にあるダウンロードマークを押して以下のファイルをダウンロードしましょう。
- ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502/ALL.chr14.phase3_shapeit2_mvncall_integrated_v5a.20130502.genotypes.vcf.gz
ブラウザによってはアドレスバーに上記のFTPアドレスを直接指定してもダウンロード可能なものもあります。Linuxのcurlコマンドを使っても構いません。
これでVCF形式のファイルがgzip形式で圧縮された状態で取得できました。以下の操作では解凍せずにそのままで解析可能です。もし解凍する場合はLinuxのターミナルからgunzipコマンドで解凍するか、7-zipなどの圧縮・解凍アプリを用いて解凍しましょう。ただし、ダウンロードサイズは500MB程度ですが解凍すると26GBを超えるので注意してください。
※ Microsoft Storeに登録されているのは公式版ではありませんが、GNU LGPLのライセンスのもとでオープンソースで公開されています。
ちなみに先ほど「VCF形式の基本構造」の項目でお示ししたサンプルデータは、ここで取得したVCFファイルの一部になります。
Pythonでバリアントコールファイルを扱うモジュール
PythonでVCF形式のファイルを扱うモジュールはいくつか存在しますが、ここではPyVCFを用いて説明します。

なお、PyVCFはVCF形式のファイルに対する基本的な操作を行う上で機能的な不足はありませんが、実行速度が遅いという欠点があります。
VCFファイルから基本情報を抽出する
VCF形式のデータを読み込む
PyVCFではVCF形式のファイルをReaderオブジェクトとして読み込みます。まずは先ほど取得したサンプルファイル(ALL.chr14.phase3_shapeit2_mvncall_integrated_v5a.20130502.genotypes.vcf.gz)をvcf_fileという名前のReaderオブジェクトとして読み込んでメタデータを表示してみましょう(ファイルパスはご自分の環境に合わせて変更してください)。なお、gzip形式で圧縮されたファイルをそのまま指定して構いません。
import vcf
vcf_file = vcf.Reader(filename='ALL.chr14.phase3_shapeit2_mvncall_integrated_v5a.20130502.genotypes.vcf.gz')
print(vcf_file.metadata)
OrderedDict([('fileformat', 'VCFv4.1'), ('fileDate', '20150218'), ('reference', 'ftp://ftp.1000genomes.ebi.ac.uk//vol1/ftp/technical/reference/phase2_reference_assembly_sequence/hs37d5.fa.gz'), ('source', ['1000GenomesPhase3Pipeline'])])
VCFファイルのメタデータが順序付き辞書(OrderedDict)で得られました。
バリアントレベルで格納されている情報を確認する
バリアントごとに格納されている付加情報(INFO列)はinfos属性で順序付き辞書(OrderedDict)として得られます。ここでは、そのKeyのみを抽出してみましょう。
import vcf
vcf_file = vcf.Reader(filename='ALL.chr14.phase3_shapeit2_mvncall_integrated_v5a.20130502.genotypes.vcf.gz')
for info_key in vcf_file.infos:
print(info_key)
CIEND CIPOS CS END IMPRECISE MC MEINFO MEND MLEN MSTART SVLEN SVTYPE TSD AC AF NS AN EAS_AF EUR_AF AFR_AF AMR_AF SAS_AF DP AA VT EX_TARGET MULTI_ALLELIC
AC、AF、NS、AN、DP、AAのようなVCF形式であらかじめ定義された項目に加えて、CIENDやCIPOSなどの独自に定義されたKeyも格納されていることが分かります。
サンプルレベルで格納されている情報を確認する
サンプルごとに格納されている情報のフォーマットはformats属性で順序付き辞書(OrderedDict)として得られます。ここでは、そのKeyのみを抽出してみましょう。
import vcf
vcf_file = vcf.Reader(filename='ALL.chr14.phase3_shapeit2_mvncall_integrated_v5a.20130502.genotypes.vcf.gz')
for format_key in vcf_file.formats:
print(format_key)
GT
各サンプルについてはGT(Genotype:遺伝子型)の情報が格納されていることが分かりました。
各バリアントについての情報を抽出する
VCF形式のデータ領域の1つのレコードが1つのバリアントに対応しています。Readerオブジェクトのデータ領域は_Recordオブジェクトをアイテムとするイテレータとして格納されているので、そのレコードを取り出してみましょう。
まずは、VCFファイルに格納されている情報をそのまま表示させます。
import vcf
vcf_file = vcf.Reader(filename='ALL.chr14.phase3_shapeit2_mvncall_integrated_v5a.20130502.genotypes.vcf.gz')
record = next(vcf_file)
print('染色体番号:' + record.CHROM)
print('変異のある塩基の位置:' + str(record.POS))
print('変異のID:' + record.ID)
print('変異部位のリファレンスゲノム上での塩基配列:' + record.REF)
print('変異部位のシークエンスデータ上での塩基配列:' + str(record.ALT))
print('変異部位のマッピングクオリティ値:' + str(record.QUAL))
染色体番号:14 変異のある塩基の位置:19000017 変異のID:rs375700886 変異部位のリファレンスゲノム上での塩基配列:C 変異部位のシークエンスデータ上での塩基配列:[T] 変異部位のマッピングクオリティ値:100
そのバリアントのタイプやサブタイプを表示させることもできます。
import vcf
vcf_file = vcf.Reader(filename='ALL.chr14.phase3_shapeit2_mvncall_integrated_v5a.20130502.genotypes.vcf.gz')
print('バリアントのタイプ:' + record.var_type)
print('バリアントのサブタイプ:' + record.var_subtype)
バリアントのタイプ:snp バリアントのサブタイプ:ts
バリアントごとの各サンプルの変異についての情報はsamples属性にサンプルを表す_Callオブジェクトのリストとして格納されています。
import vcf
vcf_file = vcf.Reader(filename='ALL.chr14.phase3_shapeit2_mvncall_integrated_v5a.20130502.genotypes.vcf.gz')
record = next(vcf_file)
samples = record.samples
print('サンプルの総数:' + str(len(samples)))
print('1番目のサンプルを表示:' + str(samples[0]))
サンプルの総数:2504 1番目のサンプルを表示:Call(sample=HG00096, CallData(GT=0|0))








コメント