がんゲノムデータベースのGDC Data Portal (TCGAデータベース)には様々な癌種の臨床情報からゲノムデータまでが格納されています。純粋なドライな研究としてもウェットの研究の前段階としても、このGDC Data Portalを用いたドライ解析を行うことは非常に重要です。ここではGDC Data Portalを用いてRNA-Seqの遺伝子発現量データを取得し、解析しやすい形に整形してその後の解析につなげるまでを説明していきます。
なお、この記事はバイオインフォマティクス Advent Calendar 2020の23日目の記事として公開しています(公開が遅くなり大変申し訳ありません)。
動作確認環境
OS
- Windows10
- Windows10 + WSL (WindowsユーザーのためのPythonを用いたゲノム解析環境)
Python
- Python 3.7.9
GDC Data Portalとは?
NIH(アメリカ国立衛生研究所)によるがんゲノムプロジェクトで、さまざまながん種について、ゲノムやエピゲノム、トランスクリプトーム、変異情報などのデータを集約して、公開しています。もともと、The Cancer Genome Atlas (TCGA)として広く知られていましたが、現在ではGDC Data Portalの一部として公開されています。
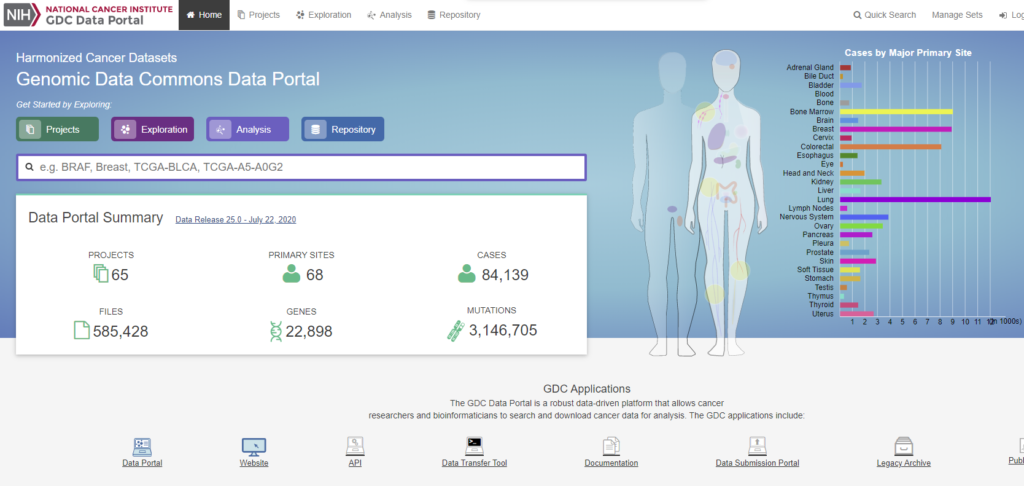
GDC Data PortalにはRNA-Seqの遺伝子発現量カウントも公開されており、この結果を用いて様々な解析を行うことが可能です。
GDC Data Portalからのデータの取得方法は以下の記事もご覧ください。

GDC Data Portalから遺伝子発現量カウントのデータを取得する
ここではGDC Data PortalからRNA-Seqの遺伝子発現量カウントのデータを取得し、その後の解析をしやすいように加工する方法を説明していきます。
条件を指定してFPKM値を取得する
ここでは、35歳未満の若年乳がん症例の遺伝子発現定量値を取得してみましょう。GDC Data Portalにおける遺伝子発現量カウントはHTSeqが主に用いられており、データベースにはカウント値は「HTSeq – Counts」、正規化したものとしては「HTSeq-FPKM」が格納されています。
それでは、以下のページの左側の条件指定欄で以下のように指定します。
条件指定
- Files
- Experimantal Strategy → RNA-Seq
- Workflow Type → HTSeq – FPKM
- Cases
- Primary Site → breast
- Age at Diagnosis → 0-34
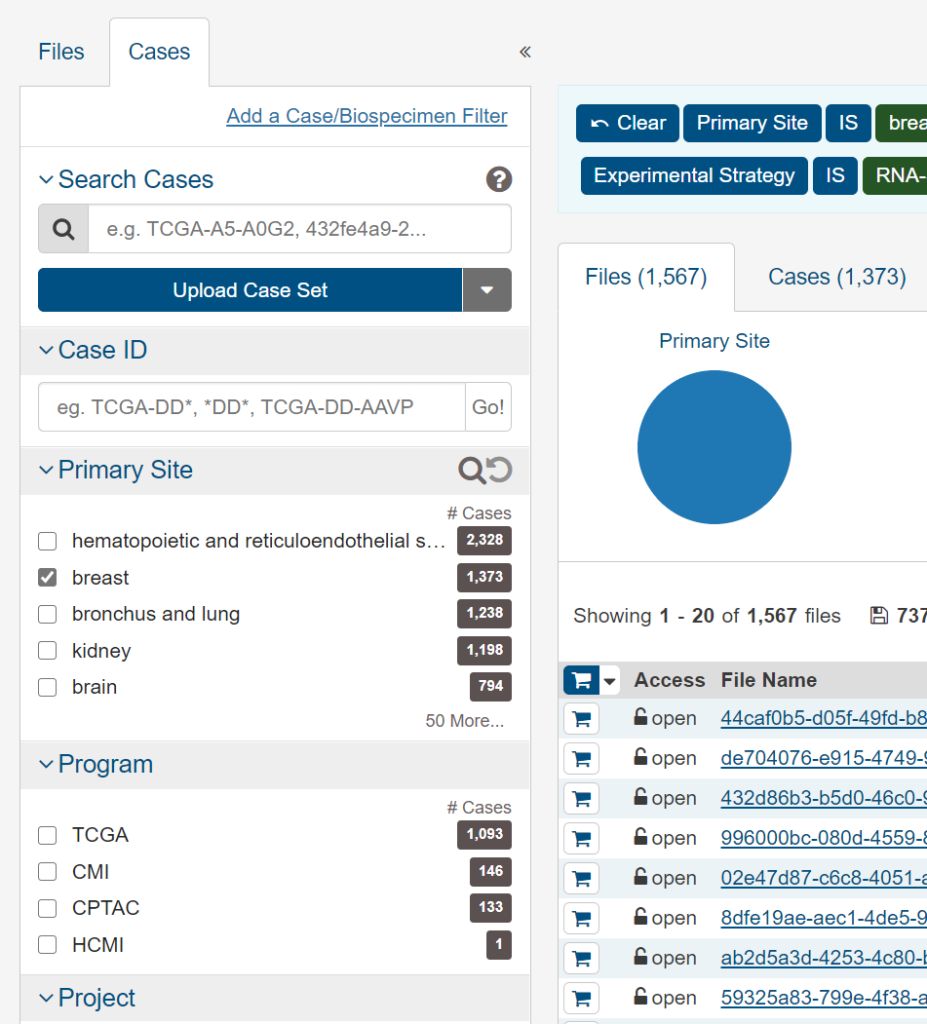
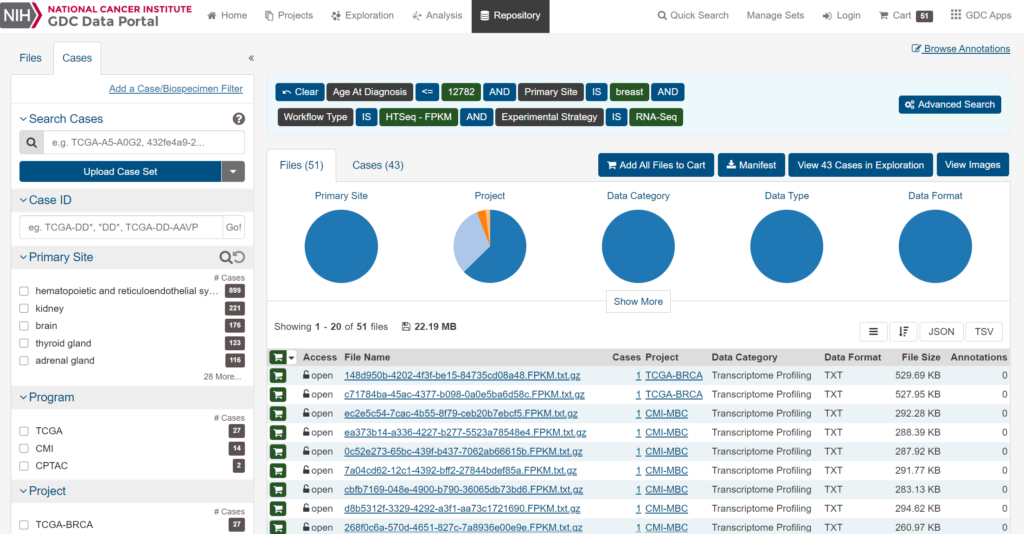
これで若年乳がん症例(35歳未満)のすべての遺伝子発現情報が抽出できましたので、「Add All Files to Cart」でカートに入れたうえで、カートからダウンロードしてください。
ダウンロードしたデータを解凍してDataFrameに格納する
ここでダウンロードしたデータは、複数のフォルダをTARで束ねた上でGZIP形式で圧縮されています。これを解凍して、後から解析しやすいように遺伝子発現量カウントのデータを1つのDataFrameにまとめましょう。
ダウンロードしたファイル名をgdc_download_xxxxxxxxxxx.tar.gzとして、そこからすべてのサンプルのFPKM値をDataFrameに格納するコードは以下のようになります。
import os
import tarfile
import pandas as pd
import shutil
import copy
# ファイル名は適宜変更してください。
with tarfile.open("gdc_download_xxxxxxxxxxx.tar.gz") as tar:
tar.extractall('tmp') # 一時フォルダを作成して、圧縮したファイルを展開する
global df # 作成するDataFrameをグローバル変数として準備しておく
counter = 0
for file_name in tar.getnames():
# annotations.txtやMANIFEST.txtを除外します
if file_name.endswith('.FPKM.txt.gz'):
# FPKM値をDataFrameとして読み込む
df_tmp = pd.read_table('tmp/' + file_name, header=None, names=['gene', os.path.split(file_name)[0]], index_col='gene')
if counter == 0:
df = copy.deepcopy(df_tmp) # 最初のFPKM値のDataFrameをコピーする
else:
df = df.join(df_tmp) # 1つ前のDataFrameに新しいDataFrameを追加していく
counter += 1
shutil.rmtree('tmp') # 一時フォルダを削除する
print(df.head())
002aa584-7dd3-4c62-952b-b24e5858482b ... fed55ba2-8a4b-4466-b176-222699cc24c1 gene ... ENSG00000242268.2 0.000000 ... 0.000000 ENSG00000270112.3 0.003032 ... 0.010501 ENSG00000167578.15 3.022233 ... 2.565837 ENSG00000273842.1 0.000000 ... 0.000000 ENSG00000078237.5 3.089273 ... 1.710800
これで次のような形式のGDC Data Portalから取得したFPKM値を次のようなDataFrameとして格納することができました。DataFrameにおけるサンプル名はFile IDとしています。

FPKM値をTPM値に変換する
GDC Data Portalに登録されているデータは先ほど取得したようにFPKM値ですが、FPKM値はサンプル間の発現量の比較に問題があることが言われており、最近ではそれを改良したTPM値が主に用いられています。

そこで、先ほどのコードを改良して、FPKM値ではなくTPM値を取得するようにしてみましょう。22-24行目でFPKMの総リード数を100万にそろえてからDataFrameに格納するように変更しています。
import os
import tarfile
import pandas as pd
import shutil
import copy
# ファイル名は適宜変更してください。
with tarfile.open("gdc_download_xxxxxxxxxxx.tar.gz") as tar:
tar.extractall('tmp') # 一時フォルダを作成して、圧縮したファイルを展開する
global df # 作成するDataFrameをグローバル変数として準備しておく
counter = 0
for file_name in tar.getnames():
# annotations.txtやMANIFEST.txtを除外します
if file_name.endswith('.FPKM.txt.gz'):
# FPKM値をDataFrameとして読み込む
df_tmp = pd.read_table('tmp/' + file_name, header=None, names=['gene', os.path.split(file_name)[0]], index_col='gene')
# FPKM値からTPM値に変換する
fpkm_sum = df_tmp.sum()[0]
df_tmp = df_tmp * 1000000 / fpkm_sum
if counter == 0:
df = copy.deepcopy(df_tmp) # 最初のFPKM値のDataFrameをコピーする
else:
df = df.join(df_tmp) # 1つ前のDataFrameに新しいDataFrameを追加していく
counter += 1
shutil.rmtree('tmp') # 一時フォルダを削除する
print(df.head())
002aa584-7dd3-4c62-952b-b24e5858482b ... fed55ba2-8a4b-4466-b176-222699cc24c1 gene ... ENSG00000242268.2 0.000000 ... 0.000000 ENSG00000270112.3 0.010348 ... 0.036526 ENSG00000167578.15 10.315710 ... 8.925140 ENSG00000273842.1 0.000000 ... 0.000000 ENSG00000078237.5 10.544537 ... 5.950935
遺伝子発現量データの解析
注目している集団の遺伝子発現量をDataFrameとして取得できたら、その遺伝子発現量の解析を行いましょう。MAプロットによる解析やクラスター解析、そして発現変動遺伝子の抽出など様々な解析がありますが、それらについては別の記事で説明する予定です。








コメント
コメント一覧 (1件)
わかりやすく例示いただき大変ありがとうございます。
実際にTCGAよりデータをダウンロードし、実施を試みましたが、エラーメッセージが出ましたので修正方法をお教えいただきますと幸です。よろしくお願いいたします。
実施コマンド:
with tarfile.open(“gdc_download_20230418_085906.562580.tar.gz”) as tar:
tar.extractall(‘tmp’)
global df
counter = 0
for file_name in tar.getnames():
if file_name.endswith(‘.FPKM.txt.gz’):
df_tmp = pd.read_table(‘tmp/’ + file_name, header=None, names=[‘gene’, os.path.split(file_name)[0]], index_col=’gene’)
fpkm_sum = df_tmp.sum()[0]
df_tmp = df_tmp * 1000000 / fpkm_sum
if counter == 0:
df = copy.deepcopy(df_tmp)
else:
df = df.join(df_tmp)
counter += 1
shutil.rmtree(‘tmp’)
print(df.head())
df.to_csv(“TCGA_CCC.csv”)
エラーメッセージ:
Traceback (most recent call last):
File “”, line 23, in
NameError: name ‘df’ is not defined