ここでは、「WindowsユーザーのためのPythonを用いたゲノム解析環境」で構築した環境で、ゲノムデータの取得方法を説明します。
バイオインフォマティクス環境
以下の方法で構築した環境であることを前提として説明します。
- Windows10 + WSL (WindowsユーザーのためのPythonを用いたゲノム解析環境)
※ 検証はしていませんが、プログラム自体は基本的にmacOS、Linuxでも動作すると思われます。
[toc]FTPでダウンロードする
まずは基本となるFTPダウンロードの方法について説明します。LinuxでFTPからダウンロードする場合は、次のコマンドを用います。
curl [オプション] [URL]
ゲノムデータの保存先のURLが分かればcurlコマンドを用いてFTPでダウンロードすることができます。
例えば、ヒトゲノムのリファレンス配列(GRCh38)は
curl -O ftp://ftp.ensembl.org/pub/current_fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna.toplevel.fa.gz
で取得できます。
それではWindows + WSL環境ではどのようにしたらよいでしょうか?
Linuxターミナルにコマンドを入力する
Visual Studio CodeのLinuxターミナルに上記のコマンドを入力してみましょう。
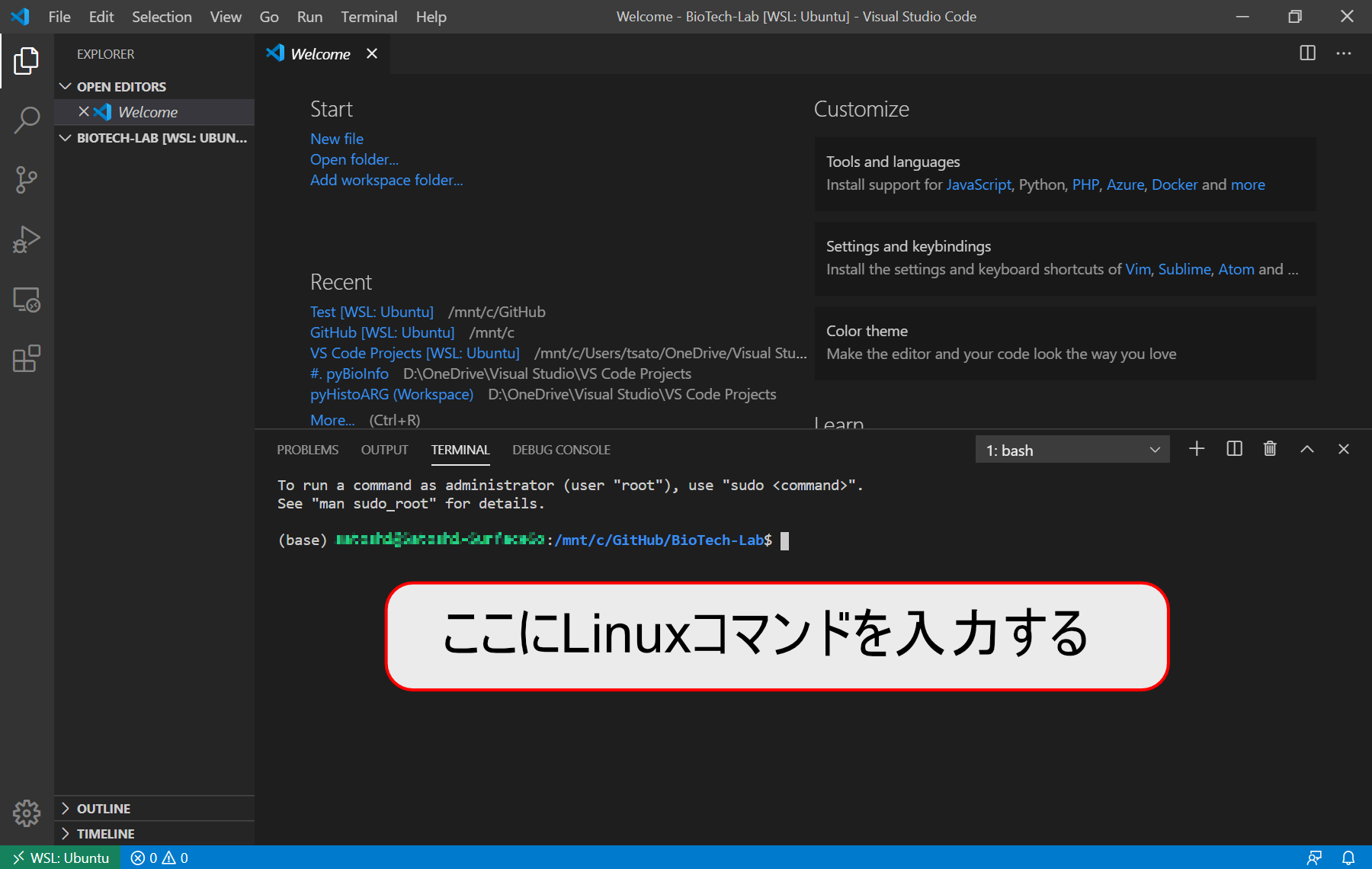
curl -O ftp://ftp.ensembl.org/pub/current_fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna.toplevel.fa.gz
Linuxターミナルの作業ディレクトリはVisual Studio Codeのワークスペースになっているので、このコマンドで自動的にワークスペースフォルダにダウンロードファイルが保存されます。
このファイルは以下のコマンドで解凍できます。
gunzip Homo_sapiens.GRCh38.dna.toplevel.fa.gz
PythonからLinuxコマンドを実行する
LinuxコマンドはPythonのsubprocess.run関数から実行することも可能です。引数にLinuxコマンドの要素を順番に文字列としてリストとして与えます。
import subprocess
proc = subprocess.run(['curl', '-O', 'ftp://ftp.ensembl.org/pub/current_fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna.toplevel.fa.gz'])
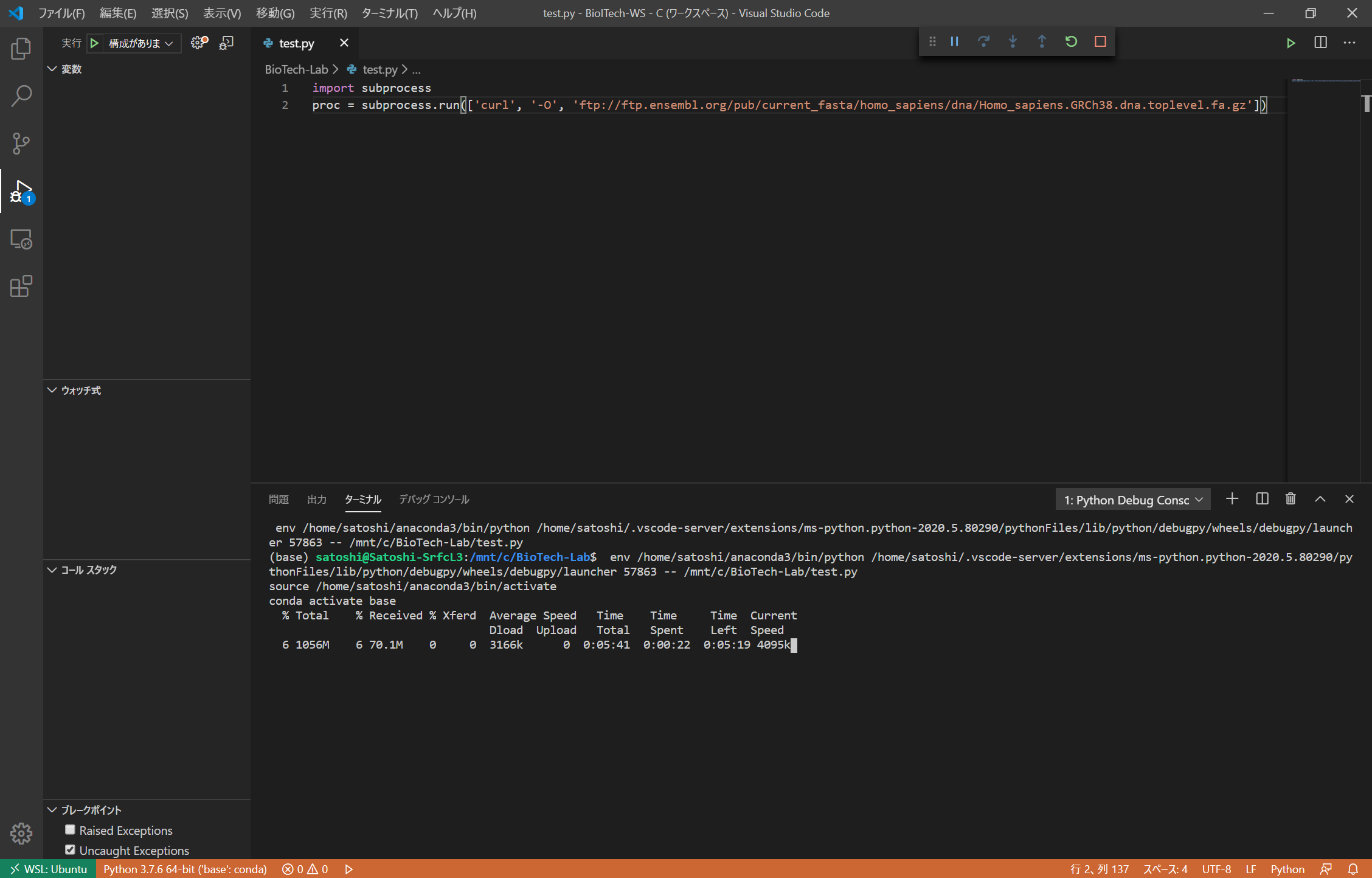
この方法を使えば、複数のファイルを一括してダウンロードするするようなスクリプトもPythonを用いて書くことができます。なお、通常のPythonスクリプトで実行するとcurlコマンドの進捗状況も上のキャプチャのように表示されますが、Jupyter Notebookを用いて実行すると進捗状況は表示されないようです。
また、取得したファイルの解凍は以下のようになります。
proc = subprocess.run(['gunzip', 'Homo_sapiens.GRCh38.dna.toplevel.fa.gz'])
PythonスクリプトでFTPダウンロードする
最後に、完全にLinuxのコマンドを使わずにPythonのみで実装してみましょう。
import urllib.request
url = 'ftp://ftp.ensembl.org/pub/current_fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna.toplevel.fa.gz'
urllib.request.urlretrieve(url, 'Homo_sapiens.GRCh38.dna.toplevel.fa.gz')
これでLinuxのコマンドを完全に排除することができました。ただし、Linuxのcurlコマンドではダウンロードの経過時間や残り時間の進捗状況もターミナルに表示されていましたが、この方法では進捗状況を表示させるためにはその処理もコーディングする必要があります。
取得したファイルの解凍は以下のようになります。
import os, gzip
with gzip.open('Homo_sapiens.GRCh38.dna.toplevel.fa.gz', 'rt') as gz_file:
with open('Homo_sapiens.GRCh38.dna.toplevel.fa', mode='w') as fasta_file:
fasta_file.write(gz_file.read())
os.remove('Homo_sapiens.GRCh38.dna.toplevel.fa.gz')
解凍処理もPythonのみでコーディング可能ですが、やや煩雑になります。
以上のように、ゲノムデータのFTPダウンロードの方法を説明しました。このサイトではPythonスクリプトに組み込んでいくことも考慮して、PythonからLinuxコマンドを実行する方法を採用していきます。
また、基本的にFTPダウンロードですべてのゲノムデータが取得可能ですが、一部のデータベースには特殊なアクセス方法が用意されていますので、その取得方法を以下にお示しします。
公共データベースからの配列データの取得
公共データベースから配列データをBiopythonを用いて取得する方法をお示しします。なお、Biopythonでは配列データはSeqRecordオブジェクトとして扱うことが基本となります。SeqRecordオブジェクトについては以下をご参照ください。

TogoWS経由でのデータ取得
TogoWSは国内主要拠点のデータベース(DDBJ、PDBj、KEGG)や海外の主要拠点データベース(NCBI、EBI, UCSC)に対する基本的な操作について、統一的なウェブサービスのインターフェイスを提供するサービスです。
以下のようなデータベースに対応しています。
- KEGG : compound, drug, enzyme, genes, glycan, orthology, reaction, module, pathway
- DDBJ : ddbj, dad, pdb
- NCBI : nuccore, nucest, nucgss, nucleotide, protein, gene, onim, homologue, snp, mesh, pubmed
- EBI : embl, uniprot, uniparc, uniref100, uniref90, uniref50
TogoWSを用いて、以下のことが可能です。TogoWSのAPIはREST APIとなっておりURLで指定しますが、ここではBiopythonのTogoWSモジュールを用います。
データベースのエントリーの取得
データを取得するデータベース名と、データのアクセッション番号、フォーマットを指定してダウンロードします。そのデータベースのデフォルトのデータ形式(ここでは、GenBank flat file)としてデータを取得して、SeqRecordオブジェクトに変換する例をお示しします。(TogoWS.entry関数を使用します)
from Bio import TogoWS, SeqIO
with TogoWS.entry('nucleotide', 'NC_045512.2') as handle:
record = SeqIO.read(handle, "genbank")
print(record)
ID: NC_045512.2
Name: NC_045512
Description: Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, complete genome
Database cross-references: BioProject:PRJNA485481
Number of features: 57
......
Seq('ATTAAAGGTTTATACCTTCCCAGGTAACAAACCAACCAACTTTCGATCTCTTGT...AAA', IUPACAmbiguousDNA())
ここで取得したデータをFASTA形式でファイルに保存する場合は次のようにします。
from Bio import TogoWS, SeqIO
with TogoWS.entry('nucleotide', 'NC_045512.2') as handle:
record = SeqIO.read(handle, "genbank")
SeqIO.write(record, 'seq.fasta', 'fasta')
なお、以下のようにしてSeqRecordオブジェクトに変換せずに直接ファイルに保存することもできます。
from Bio import TogoWS
with TogoWS.entry('nucleotide', 'NC_045512.2', 'fasta') as handle:
with open('seq2.fasta', 'w') as f:
f.write(handle.read())
データベースにおいて、与えられた条件を満たすデータの検索
nucleotideのデータベースから’lung cancer’に関するものを検索して、1番目から5番目までの結果を表示します。(TogoWS.search関数を使用します)
from Bio import TogoWS
result = TogoWS.search('nucleotide', 'lung cancer', 1, 5)
print(result.read())
1842337669 1842280863 1831751460 1831751452 1819861774
ここで得られたIDを次のようにentry関数のidに指定することで、そのデータを取得することができます。
TogoWS.entry('nucleotide', '1842337669', 'gb')
データファイルの形式を変換する
TogoWSを通してGenBankのデータを取得し、それをFASTAに変換する例をお示しします。(TogoWS.convert関数を使用します)
from Bio import TogoWS
with TogoWS.entry('nucleotide', 'NC_045512.2') as handle:
with TogoWS.convert(handle, 'genbank', 'fasta') as handle_fasta:
print(handle_fasta.read())
この結果はentry関数で直接FASTAを取得するのと同じ結果になります。
from Bio import TogoWS
with TogoWS.entry('nucleotide', 'NC_045512.2', 'fasta') as handle:
print(handle.read())
Entrez経由でのNCBIデータベースからのデータ取得
TogoWSを用いてNCBIデータベースにアクセスすることもできますが、NCBIデータベースには専用のインターフェースとしてEntrezが用意されています。Entrezを用いることで、TogoWSだけではできないような複雑な検索やNCBIデータベース特有の情報を得ることができます。
Entrezの使用法については以下の記事をご覧ください。
公共データベースからのNGS出力ファイル(SRAファイル)の取得
NCBIが配布しているSRA Toolkitを用いてSRAファイルを直接ダウンロード可能です。fasterq-dumpコマンドにSRA RUN IDを指定して実行すると、直接NCBIデータベースからダウンロードして、FASTQファイルに変換してくれます。
SRA Toolkitをインストールしていない場合は、condaを使ってbiocondaからインストールしましょう。Visual Studio CodeのLinuxターミナルで次のコマンドを実行します。
conda install -c bioconda sra-tools
なお、fasterq-dumpはsra-tools 2.9.1以上でfastq-dumpの後継として登場したものなので、sra-toolsのバージョンが低い場合は以下のようにバージョン指定しましょう。
conda install -c bioconda sra-tools=2.9.1
例えばSRA RUN IDがDRR042266のものを取得する場合は、fasterq-dumpの引数にSRA RUN IDを指定するだけで取得できます。(-pオプションで進捗状況を表示します。)
fasterq-dump SRR000001 -p
それでは、fasterq-dumpをPythonのsubprocessから実行してみましょう。
import subprocess
proc = subprocess.run(['fasterq-dump', 'SRR000001', '-p'])
これでシークエンスデータをダウンロードして、作業フォルダに保存することができました。








コメント