ゲノムアノテーションファイルとしてGFF形式やGFF形式から派生したGTF形式などがあります。ここではGFF形式の構造について説明し、Pythonによる基本操作を解説します。
動作確認環境
OS
- Windows10 + WSL (WindowsユーザーのためのPythonを用いたゲノム解析環境)
Python
- Python 3.7.6
モジュール
- BCBio-GFF 0.6.6
GFF形式のバージョンとその基本構造
GFF形式の種類
GFF形式にはいくつかのバージョンがあり、現在ではversion2 (GFF2)とversion3 (GFF3)がよく使われています。また、GFF2をもとにした規格としてGTFというものもあります(規格としてはGFF2とGTFは同一)。
ここではGFF形式のうち、特にGFF3形式を扱っていきます。
GFF3形式の構造
GFF3形式はテキスト形式で、1つの行が1つのfeature領域のアノテーションを表します。また先頭が「##」で始まる行にはメタデータ、「#」で始まる行にはコメントが記載されています。なお、NCBIによるGFF3形式では「#!」もメタデータとして扱われていますが、GFF3形式の公式規則ではなくあくまでもNCBIのローカルルールとして運用されています。
例えばGRCh38の21番染色体のゲノムアノテーションを表すGFF3形式は以下のようになります。
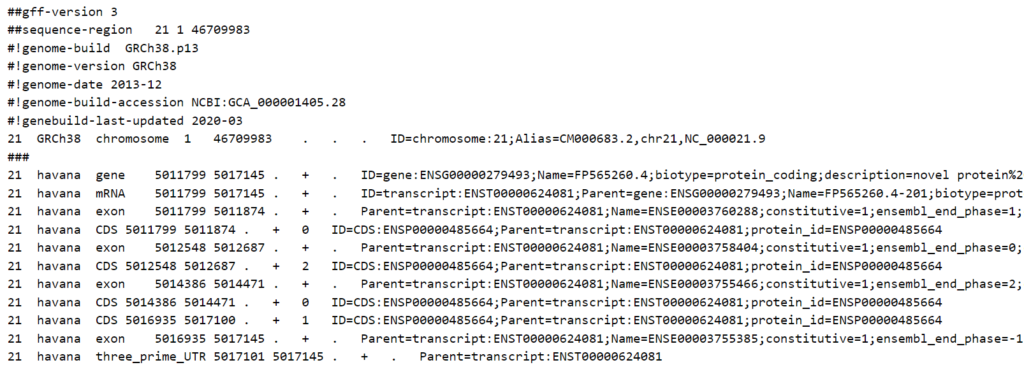
アノテーション部分の各行はタブ区切りで左から、
| seqid | そのfeature領域を含む染色体のID |
| source | アノテーションに用いたソフトウェアやデータベース |
| type | feature領域のタイプ(gene, exon, CDS, mRNAなど) |
| start | feature領域の開始位置 |
| end | feature領域の終了位置 |
| score | feature領域の信頼度スコア |
| strand | feature領域の遺伝子の向き(プラス鎖 / マイナス鎖) |
| phase | typeがCDSの時に、何番目の塩基からコドンが始まるかを表す(0 / 1 / 2) |
| attributes | feature領域の属性(tag=valueの形式で表現され、セミコロン区切りで複数の属性を指定可能) |
となります。該当する項目が空欄の場合は「.」で表記されます。
なお、attributesのtagは自由に設定可能ですが、大文字で始まるtagはあらかじめ予約されたtag名となっています。あらかじめ予約されたtagとしては以下のようなものがあります。
| ID | feature領域のID |
| Name | feature領域の名前 |
| Parent | そのfeature領域の親となる領域を表すID |
| Note | メモ |
| Dbxref | データベースの相互参照 |
| Is_circular | そのfeature領域が環状構造であるかどうか |
追加のtagとしては、例えばNCBIによるGFF3形式では「anticodon、description、exception、exon_number、gbkey、gene、gene_biotype、gene_synonym、locus_tag、ncrna_class、part、partial、product、protein_id、pseudo、pseudogene、start_range、end_range、transcript_id、transl_except」などが用意されています。詳細はNCBIのドキュメント(What is GFF3 format?)をご覧ください。
その他、GFF3形式の詳細については以下をご覧ください。
サンプルで用いるGFF3形式ファイル
ここではサンプルとしてヒトのリファレンスゲノム配列(GRCh38)の21番染色体のアノテーションファイルをGFF3形式で取得しましょう。以下のEnsemblのサイトから右側の「Gene annotation」の部分の「Download GTF or GFF3 files for genes, cDNAs, ncRNA, proteins」のGFF3をクリックします。
GRCh38のアノテーションファイルのFTPサイトが表示されるので、このうち21番染色体を表す以下のFTPアドレスからダウンロードしてください。
- ftp://ftp.ensembl.org/pub/release-101/gff3/homo_sapiens/Homo_sapiens.GRCh38.101.chromosome.21.gff3.gz
ここで取得したGFF3形式のファイルはgzip形式で圧縮されているので、7-zipなどの圧縮・解凍アプリを用いて解凍しましょう。
※ Microsoft Storeに登録されているのは公式版ではありませんが、GNU LGPLのライセンスのもとでオープンソースで公開されています。
なお、リファレンスゲノム配列については以下の記事もご参照ください。

PythonでGFF3形式ファイルを扱うモジュール
GFF3形式のファイルはBCBio-GFFモジュールから扱うことができます。なお、BCBio-GFFモジュールは将来的にBiopythonのライブラリに統合される予定となっています。
(BCBio-GFFがBiopythonに統合されたら、以下の内容をアップデートします)
BCBio-GFFモジュールは以下のコマンドでAnacondaからインストール可能です。(bcbio-gff – Anaconda Cloud)
conda install -c bioconda bcbiogff
また、BCBio-GFF以外にもHTSeqモジュールやその他のモジュールを用いてもGFF3形式のファイルを扱うことは可能です。
GFF3形式ファイルの構造を確認する
まずはこれから扱おうとしているGFF3形式ファイルの構造を確認しておきましょう。
GFF3形式のファイルにおける各項目の一覧とそのアノテーション数を表示します。
import pprint
from BCBio.GFF import GFFExaminer
gff_file = 'Homo_sapiens.GRCh38.101.chromosome.21.gff3'
with open(gff_file) as handle:
examiner = GFFExaminer()
pprint.pprint(examiner.parent_child_map(handle))
{'gff_id': {('21',): 35165},
'gff_source': {('.',): 3463,
('GRCh38',): 1,
('ensembl',): 2247,
('ensembl_havana',): 7513,
('havana',): 19124,
('havana_tagene',): 2730,
('mirbase',): 87},
'gff_source_type': {('.', 'biological_region'): 3463,
('GRCh38', 'chromosome'): 1,
(中略)
('mirbase', 'miRNA'): 29,
('mirbase', 'ncRNA_gene'): 29},
'gff_type': {('CDS',): 7899,
('V_gene_segment',): 1,
('biological_region',): 3463,
('chromosome',): 1,
('exon',): 16982,
('five_prime_UTR',): 1593,
('gene',): 262,
('lnc_RNA',): 1657,
('mRNA',): 1002,
('miRNA',): 29,
('ncRNA',): 24,
('ncRNA_gene',): 425,
('pseudogene',): 188,
('pseudogenic_transcript',): 188,
('rRNA',): 4,
('snRNA',): 21,
('snoRNA',): 15,
('three_prime_UTR',): 1383,
('unconfirmed_transcript',): 28}}
GFF3形式のアノテーションには親子関係があるので、その親子関係を表示してみます。{親項目 : [子項目1, 子項目2, … ]}で示されています。
import pprint
from BCBio.GFF import GFFExaminer
gff_file = 'Homo_sapiens.GRCh38.101.chromosome.21.gff3'
with open(gff_file) as handle:
examiner = GFFExaminer()
pprint.pprint(examiner.available_limits(handle))
{('ensembl', 'lnc_RNA'): [('ensembl', 'exon')],
('ensembl', 'mRNA'): [('ensembl', 'CDS'),
('ensembl', 'exon'),
('ensembl', 'five_prime_UTR'),
('ensembl', 'three_prime_UTR')],
('ensembl', 'ncRNA'): [('ensembl', 'exon')],
('ensembl', 'ncRNA_gene'): [('ensembl', 'ncRNA'),
('ensembl', 'rRNA'),
('ensembl', 'snRNA'),
('ensembl', 'snoRNA')],
(中略)
('havana_tagene', 'ncRNA_gene'): [('havana_tagene', 'lnc_RNA')],
('mirbase', 'miRNA'): [('mirbase', 'exon')],
('mirbase', 'ncRNA_gene'): [('mirbase', 'miRNA')]}
GFF3形式ファイルを読み込む
bcbio-gffモジュールではSeqRecordオブジェクトとしてGFF3形式のファイルを読み込むことができます。読み込まれたGFF3形式のデータはSeqRecordオブジェクトをアイテムとするイテレータとして格納され、1つの染色体(seqid)が1つのSeqRecordオブジェクトに対応します。
SeqRecordオブジェクトの属性とGFF3形式の項目は次のように対応します。
| SeqRecordオブジェクトの属性 | GFF3形式の項目 | |
|---|---|---|
| annotations | ←→ | メタデータ(「##」で始まる行) |
| description | ←→ | なし |
| dbxrefs | ←→ | なし |
| features | ←→ | アノテーション |
| id | ←→ | 染色体のID(seqid) |
| letter_annotations | ←→ | なし |
| name | ←→ | なし |
| seq | ←→ |
GFF3形式のアノテーションの本体はSeqRecordオブジェクトのfeatures属性に格納されています。このfeatures属性はSeqFeatureオブジェクトのリストであり、親子関係のあるアノテーションでは最も上位のアノテーションが抽出されて1つのアノテーションが1つのSeqFeatureオブジェクトに変換されます。それより下の階層のアノテーションは上位階層のアノテーションのsub_features属性にSeqFeatureオブジェクトとして格納されています。
SeqFeautreオブジェクトの属性とGFF3形式の項目との関係は以下のようになります。
| SeqFeatureオブジェクトの属性 | GFF3形式の項目 | |
|---|---|---|
| id | ←→ | feature領域のID |
| location | ←→ | feature領域の開始位置・終了位置 |
| qualifiers | ←→ | feature領域の属性(key, valueの組み合わせ) |
| type | ←→ | feature領域のタイプ(gene, exon, CDS, mRNAなど) |
| sub_features | ←→ | 1つ下の階層のアノテーション(子アノテーション) |
先ほど取得したサンプルファイルを読み込んで、3番目のアノテーションを表示してみましょう。GFF.parse関数は最も上位の階層のアノテーションを取得し、取得したSeqFeatureオブジェクトはfeature領域のタイプ別に並び替えられます。
from BCBio import GFF
gff_file = 'Homo_sapiens.GRCh38.101.chromosome.21.gff3'
with open(gff_file) as handle:
for record in GFF.parse(handle):
print('メタデータ')
print(record.annotations)
print('染色体番号:' + str(record.id))
print('-----------')
print(record.features[0])
メタデータ
{'gff-version': ['3'], 'sequence-region': [('21', 0, 46709983)]}
染色体番号:21
-----------
type: biological_region
location: [5020207:5023177]
qualifiers:
Key: external_name, Value: ['oe = 0.75']
Key: logic_name, Value: ['cpg']
Key: score, Value: ['2.14e+03']
この方法ではGFF形式のアノテーションの親子関係が反映されているので、下位階層の子アノテーションを取得する場合はsub_features属性を使いましょう。
親子関係を無視してすべてのアノテーションを取得する方法としてはGFF.parse関数の引数にtarget_lines=1と指定して、1行ずつ別々のSeqRecordオブジェクトとして取得する方法があります。この場合は1つのSeqRecordオブジェクトに1つのSeqFeatureオブジェクトが含まれており、アノテーションの数だけSeqRecordオブジェクトが生成されます。
from BCBio import GFF
gff_file = 'Homo_sapiens.GRCh38.101.chromosome.21.gff3'
with open(gff_file) as handle:
records = GFF.parse(handle, target_lines=1)
record = next(records)
print('メタデータ')
print(record.annotations)
print('染色体番号:' + str(record.id))
print('-----------')
print(record.features[0])
メタデータ
{}
染色体番号:21
-----------
type: chromosome
location: [0:46709983]
id: chromosome:21
qualifiers:
Key: Alias, Value: ['CM000683.2', 'chr21', 'NC_000021.9']
Key: ID, Value: ['chromosome:21']
Key: source, Value: ['GRCh38']








コメント