NGSから配列データがFASTQ形式で得られたら、まずはそのクォリティをチェックする必要があります。FastQCというアプリケーションで品質チェック・管理を行うことが一般的ですが、ここではPythonのプログラムでFastQCの品質チェックを再現してみましょう。実際に手を動かして試してみれば、FastQCの品質チェックがよく理解できて、FASTQ形式のデータの理解も深まると思います。
バイオインフォマティクス環境
OS
- Windows10 + WSL (WindowsユーザーのためのPythonを用いたゲノム解析環境)
Python
- Python 3.7.6
モジュール
- Biopython 1.76
サンプルデータ
ここではFASTQ形式のゲノムデータのサンプルとしてDDBJ DRAデータベースに最初に登録されたシークエンスランのDRR000001を用います。以下の記事を参考に、ゲノムデータを取得してください。

DRR000001の取得、解凍に必要なコマンドは以下の通りです。
curl -O ftp://ftp.ddbj.nig.ac.jp/ddbj_database/dra/fastq/DRA000/DRA000001/DRX000001/DRR000001.fastq.bz2
bunzip2 DRR000001.fastq.bz2
ちなみに、このDRR000001は慶応義塾大学による納豆菌のゲノムデータです。
FASTQ形式のデータ構造
FASTQ形式の構造についてはこちらをご覧ください。
FastQCを用いた品質チェック
一般的にNGSでFASTQ形式の配列データが得られたら、FastQCで品質のチェック・管理が行われます。実際の研究ではデファクトスタンダードとなっているFastQCを使うことになると思いますが、ここでは練習のためにPythonを使ってFastQCの品質チェックで何をやっているか実際に確かめてみましょう。
なお、WSL環境ではGUIアプリを動かせないので、以下からWindows版をダウンロードしてzipファイルを解凍して適当なフォルダに保存してください。作成されたフォルダの「run_fastqc.bat」をダブルクリックすれば起動します。
FastQCはJavaプログラムなので、Javaのインストールも必要です。FastQCがうまく起動しない場合はJavaのインストールができていない場合がありますのでご確認ください。
FastQCのレポートの詳細は以下のサイトに詳細に書かれてあり、有益な情報が多くありますのでご参照ください。
Pythonで配列データの品質を確認してみる
配列データの基本統計を確認する
まず、FastQCでDRR000001の基本統計を確認してみます。
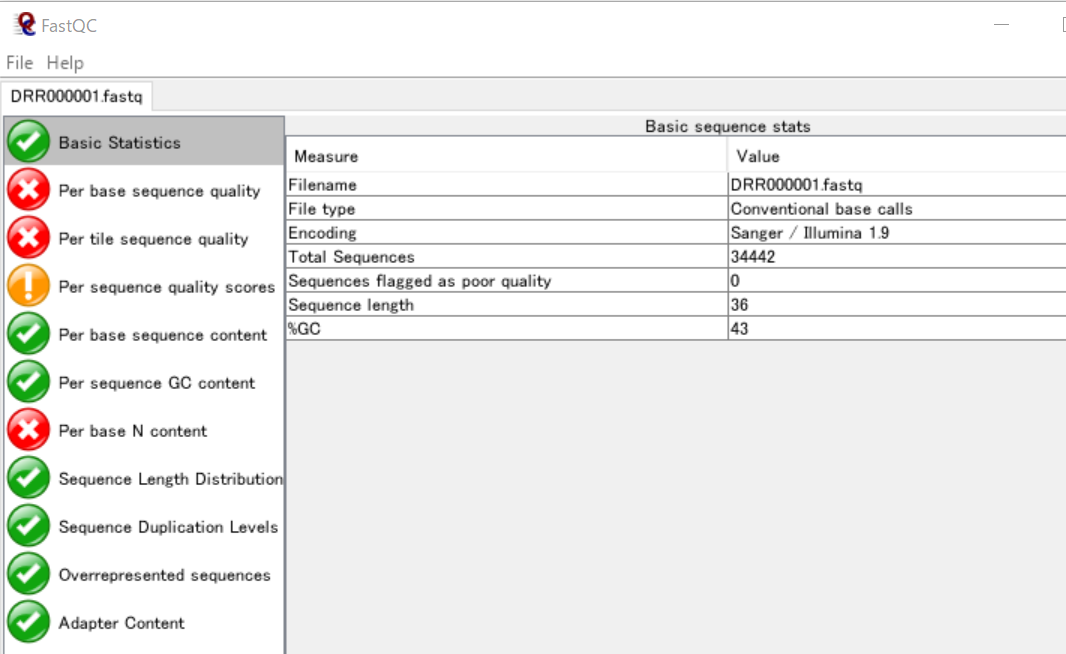
ここから、このFASTQファイルに含まれている配列は、リード数が34442で、各リードには36個の塩基が含まれていることが分かります。また、GC含量は43%です。
それでは、ここで算出されている統計情報を自分でPythonのプログラムを作って求めてみましょう。FASTQ形式のデータをSeqIO.parse関数を用いて読み込み、forループで各リードごとにSeqRecordオブジェクトとして取得して、処理をしていきます。
from Bio import SeqIO
# FASTQ形式のデータをリードごとに読み込みます
records = SeqIO.parse('DRR000001.fastq', 'fastq')
sequence_length_list = [] # リードごとの塩基数を格納するリスト
GC_count = 0 # GCの合計を格納する
ATGC_count = 0 # ATGCの合計を格納する
# リードごとに解析
for record in records:
sequence_length_list.append(len(record.seq))
GC_count += (record.seq.count('G') + record.seq.count('C'))
ATGC_count += (record.seq.count('A') + record.seq.count('T') + record.seq.count('G') + record.seq.count('C'))
print('リード数:' + str(len(sequence_length_list)))
print('リード長:' + str(min(sequence_length_list)) + '-' + str(max(sequence_length_list)))
print('GC含量:' + str(GC_count / ATGC_count * 100) + '%')
リード数:34442 リード長:36-36 GC含量:43.453617853777445%
リード中の塩基位置ごとのSequence Quality値の分布を可視化する
まず、FastQCで表示してみます。
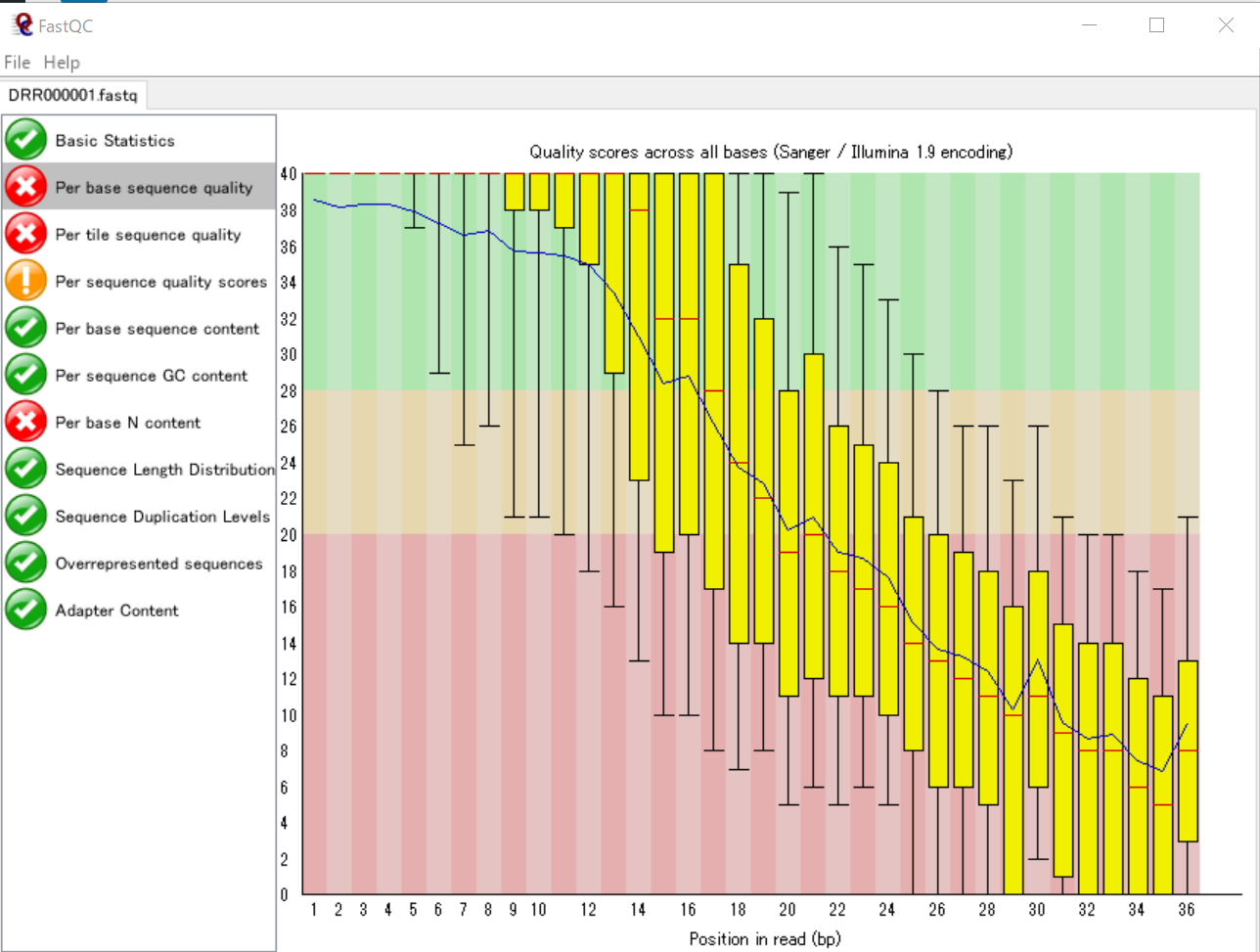
この配列データに含まれている各リードには36個の塩基が含まれており、リード内の塩基の位置ごとのSequence Quality値の分布をグラフに表しています。つまり、34442個のリードについてその「1番目の塩基のSequence Quality値の分布」「2番目の塩基のSequence Quality値の分布」…「36番目の塩基のSequence Quality値の分布」 を箱ひげ図で表したものになります。
それでは、PythonのプログラムでFASTQ形式のデータを自分で解析してみましょう。FASTQ形式のデータをSeqRecordオブジェクトに読み込むと、Sequence Quality値はletter_annotations属性にキーが’phred_quality’で、値がSequence Quality値の辞書(類似)の形式で格納されます。
なお、箱ひげ図については以下の記事もご覧ください。

from Bio import SeqIO
import matplotlib.pyplot as plt
# FASTQ形式のデータを読み込む
records = SeqIO.parse('DRR000001.fastq', 'fastq')
# リードごとの塩基数を取得する
sequence_length_list = []
for record in records:
sequence_length_list.append(len(record.seq))
# 最も塩基数が多いリードのリード長に合わせて空白のリストを生成する
quality_per_base_pos_list = []
for i in range(max(sequence_length_list)):
quality_per_base_pos_list.append([])
# recordsのイテレータを先頭に戻すために再読み込み
records = SeqIO.parse('DRR000001.fastq', 'fastq')
# 塩基位置ごとに各リードのSequence Quality値を格納する
for record in records:
for i in range(len(record.seq)):
quality_per_base_pos_list[i].append(record.letter_annotations['phred_quality'][i])
# 箱ひげ図で塩基位置ごとのSequence Quality値の分布を表示する
fig, ax = plt.subplots(figsize=(16,9))
ax.boxplot(quality_per_base_pos_list, sym='', whis=[10,90], showmeans=True)
plt.savefig('fig.png')

FastQCと同じグラフが得られました。
各リードの平均Sequence Quality値のヒストグラムを表示する
まず、FastQCで表示してみます。

各リードのSequence Quality値のヒストグラムです。
それでは、PythonのプログラムでFASTQ形式のデータを自分で解析してみましょう。
from Bio import SeqIO
import matplotlib.pyplot as plt
# FASTQ形式のデータを読み込む
records = SeqIO.parse('DRR000001.fastq', 'fastq')
# 各リードの平均Sequence Quality値をリストとして取得する
average_quality_per_sequence = []
for record in records:
average_quality = sum(record.letter_annotations['phred_quality']) / len(record.letter_annotations['phred_quality'])
average_quality_per_sequence.append(average_quality)
# 各リードの平均Sequence Quality値のヒストグラムを表示する
fig, ax = plt.subplots(figsize=(16,9))
ax.hist(average_quality_per_sequence, bins=40, range=(0, 40))
plt.savefig('fig.png')
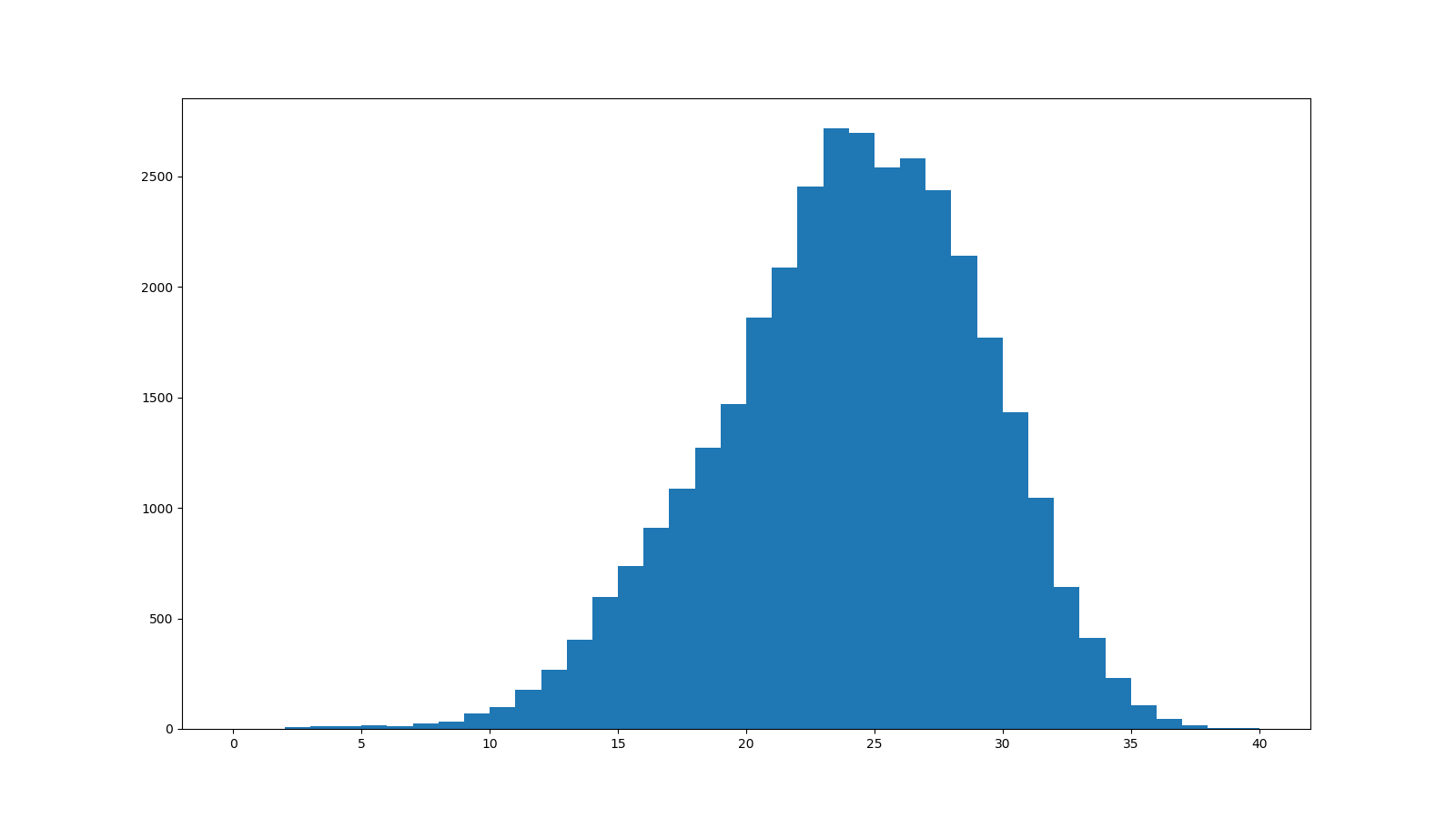
折れ線グラフと棒グラフの違いはありますが、FastQCのグラフと同じグラフが得られました。
リード中の塩基位置ごとのATGCの割合を可視化する
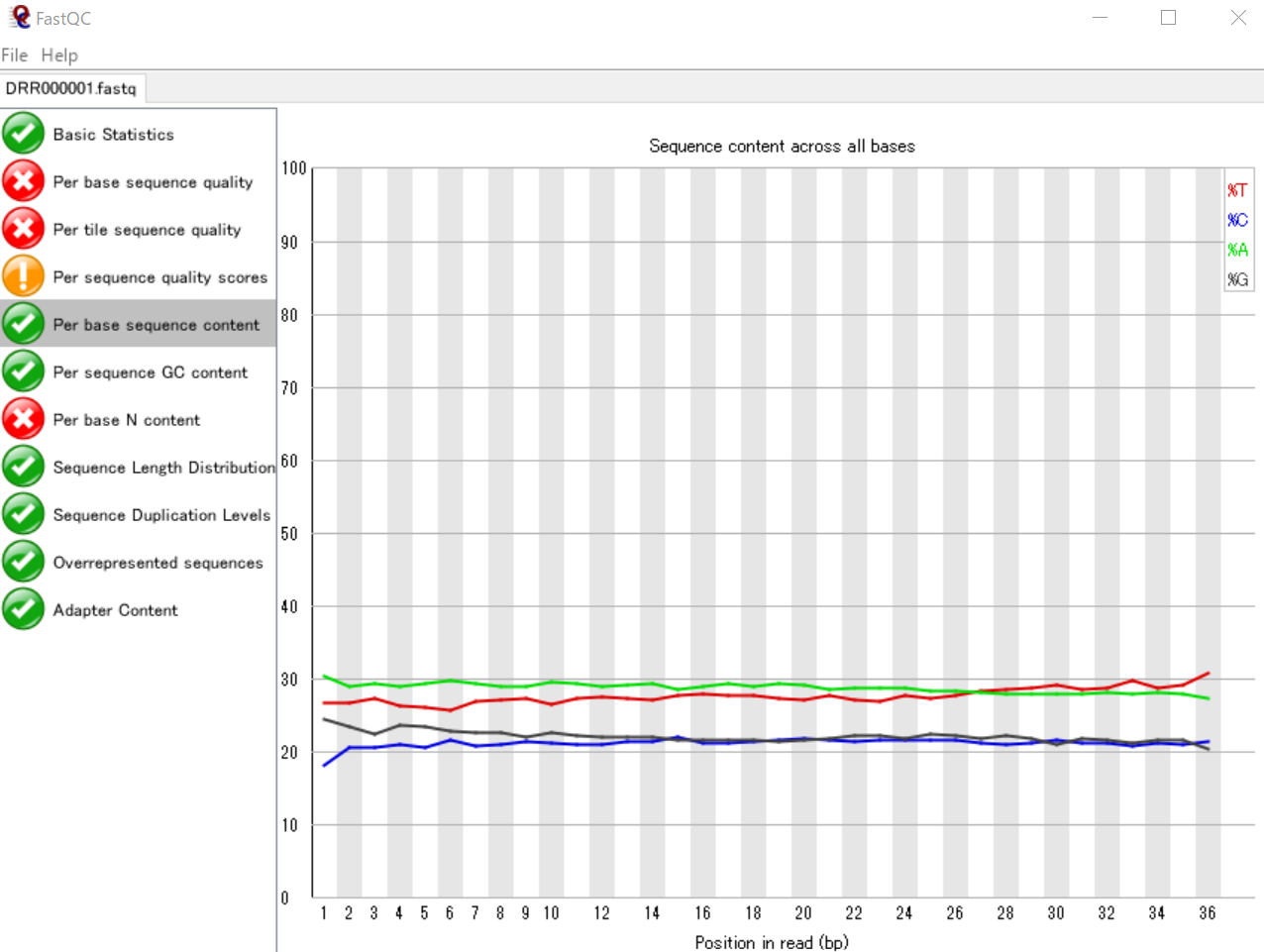
リード内のそれぞれの塩基位置(1番目~36番目)における、ATGCのそれぞれの割合を表いしています。
それでは、PythonのプログラムでFASTQ形式のデータを自分で解析してみましょう。
from Bio import SeqIO
import matplotlib.pyplot as plt
# FASTQ形式のデータを読み込む
records = SeqIO.parse('BioTech-Lab/6367/DRR000001.fastq', 'fastq')
# リードごとの塩基数を取得する
sequence_length_list = []
for record in records:
sequence_length_list.append(len(record.seq))
# 最も塩基数が多いリードのリード長に合わせて空白のリストを生成する
ATGC_per_base_pos_list = []
for i in range(max(sequence_length_list)):
ATGC_per_base_pos_list.append([])
# recordsのイテレータを先頭に戻すために再読み込み
records = SeqIO.parse('BioTech-Lab/6367/DRR000001.fastq', 'fastq')
# 各リードの位置ごとの塩基のリストを作成する
for record in records:
for i in range(len(record.seq)):
ATGC_per_base_pos_list[i].append(record.seq[i])
# リード内の位置ごとの各塩基の含まれる割合のリストを作成する
A_ratio = []
T_ratio = []
G_ratio = []
C_ratio = []
for ATGC_per_base_pos in ATGC_per_base_pos_list:
A_count = ATGC_per_base_pos.count('A')
T_count = ATGC_per_base_pos.count('T')
G_count = ATGC_per_base_pos.count('G')
C_count = ATGC_per_base_pos.count('C')
ALL_count = A_count + T_count + G_count + C_count
A_ratio.append(A_count / ALL_count * 100)
T_ratio.append(T_count / ALL_count * 100)
G_ratio.append(G_count / ALL_count * 100)
C_ratio.append(C_count / ALL_count * 100)
# リード内の塩基の位置ごとのATGCの割合を折れ線グラフで表示する
fig, ax = plt.subplots(figsize=(16,9))
ax.plot(A_ratio, label='A(%)')
ax.plot(T_ratio, label='T(%)')
ax.plot(G_ratio, label='G(%)')
ax.plot(C_ratio, label='C(%)')
ax.legend()
plt.savefig('fig.png')
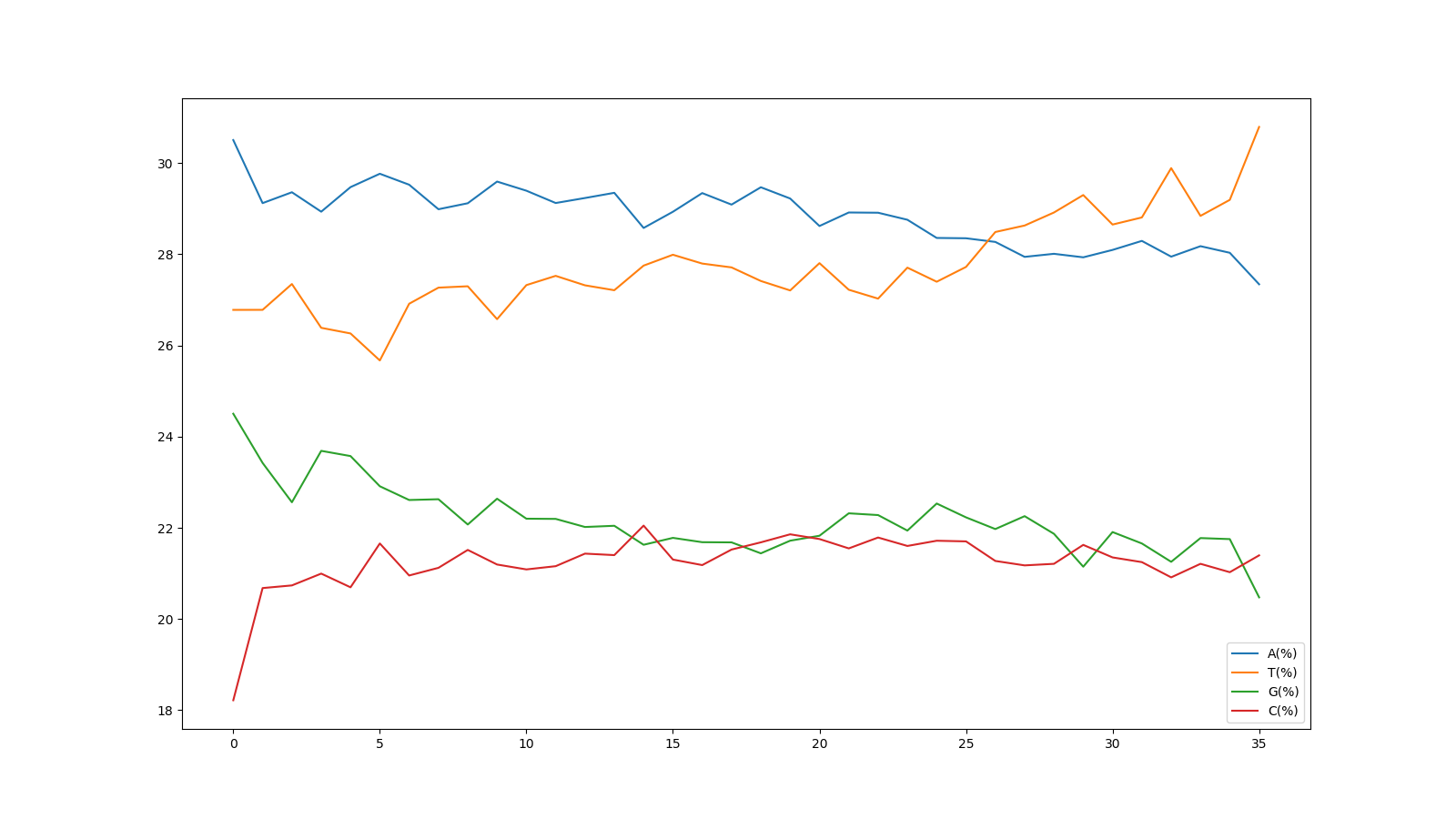
縦軸の表示範囲は18~30%となっていますが、FastQCのグラフと同じものが得られました。
各リードのGC含量のヒストグラムを表示する

各リードのGC含量をヒストグラムで表しています。それではPythonを用いてこれを再現してみましょう。
from Bio import SeqIO
import matplotlib.pyplot as plt
# FASTQ形式のデータを読み込む
records = SeqIO.parse('DRR000001.fastq', 'fastq')
# 各リードのGC含量をリストとして取得する
GC_ratio_per_sequence = []
for record in records:
GC_count = record.seq.count('G') + record.seq.count('C')
ATGC_count = GC_count + record.seq.count('A') + record.seq.count('T')
GC_ratio = GC_count / ATGC_count * 100
GC_ratio_per_sequence.append(GC_ratio)
# 各リードのGC含量のヒストグラムを表示する
fig, ax = plt.subplots(figsize=(16,9))
ax.hist(GC_ratio_per_sequence, bins=35, range=(0, 100))
plt.savefig('fig.png')

FastQCのグラフとは若干違いますが、ヒストグラムのビンの取り方による違いだと思われます。
リード中の塩基位置ごとに読み取りに失敗した塩基数の割合を可視化する
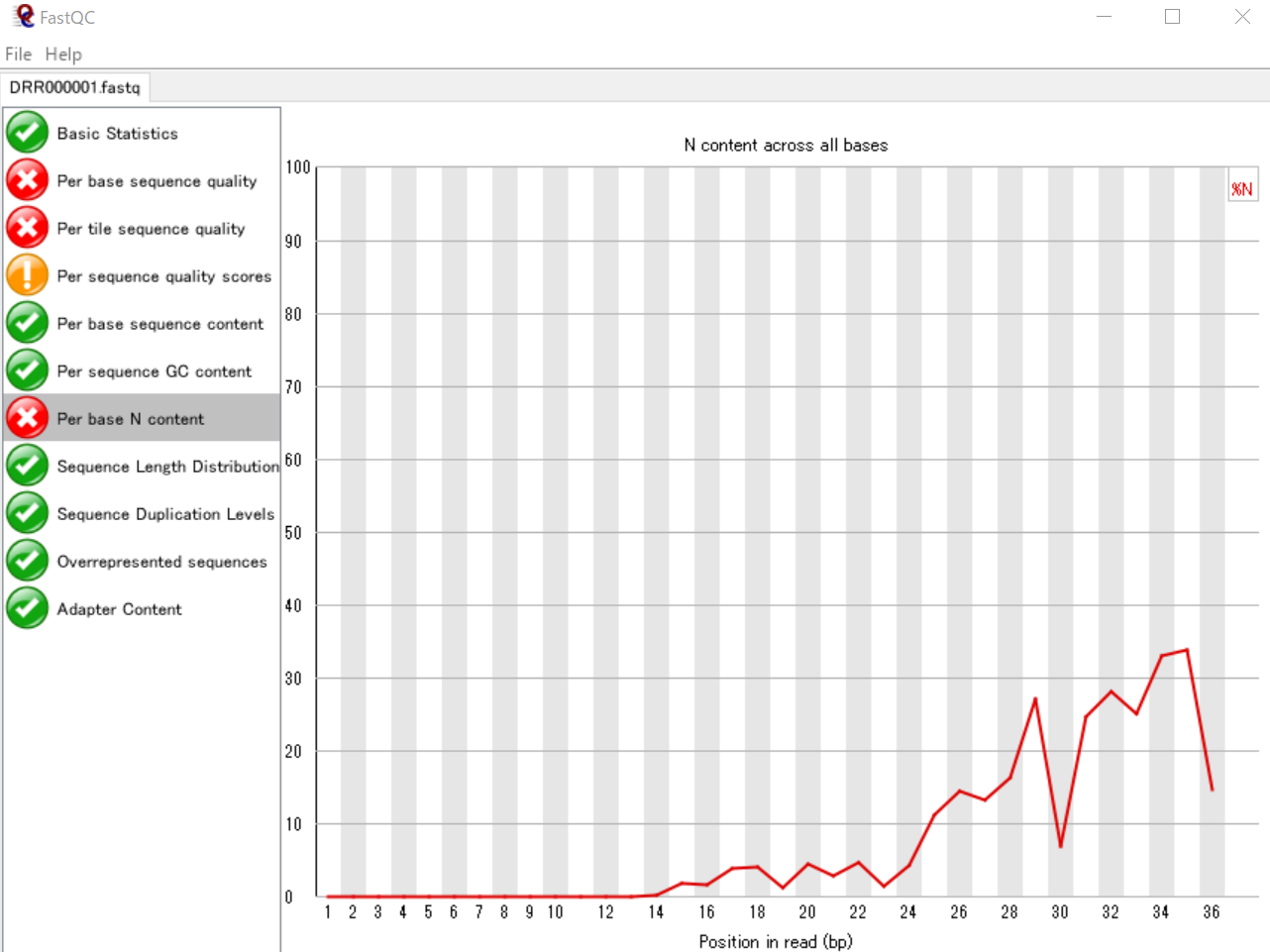
NGSの配列データでは、読み取りに失敗した塩基は「N」で表されます。したがって、ATGCの代わりにNとなっている塩基が多いと読み取りに失敗してる割合が高いといえます。ここでは、各リードの塩基位置ごとのNの割合を表しています。
これをPythonを用いて再現すると以下のようになります。
from Bio import SeqIO
import matplotlib.pyplot as plt
# FASTQ形式のデータを読み込む
records = SeqIO.parse('DRR000001.fastq', 'fastq')
# リードごとの塩基数を取得する
sequence_length_list = []
for record in records:
sequence_length_list.append(len(record.seq))
# 最も塩基数が多いリードのリード長に合わせて空白のリストを生成する
ATGCN_per_base_pos_list = []
for i in range(max(sequence_length_list)):
ATGCN_per_base_pos_list.append([])
# recordsのイテレータを先頭に戻すために再読み込み
records = SeqIO.parse('DRR000001.fastq', 'fastq')
# 各リードの位置ごとの塩基のリストを作成する
for record in records:
for i in range(len(record.seq)):
ATGCN_per_base_pos_list[i].append(record.seq[i])
# リード内の位置ごとのNの含まれる割合のリストを作成する
N_ratio = []
for ATGCN_per_base_pos in ATGCN_per_base_pos_list:
N_count = ATGCN_per_base_pos.count('N')
N_ratio.append(N_count / len(ATGCN_per_base_pos) * 100)
# リード内の塩基の位置ごとのNの割合を折れ線グラフで表示する
fig, ax = plt.subplots(figsize=(16,9))
ax.plot(N_ratio, label='N(%)')
ax.legend()
plt.savefig('fig.png')
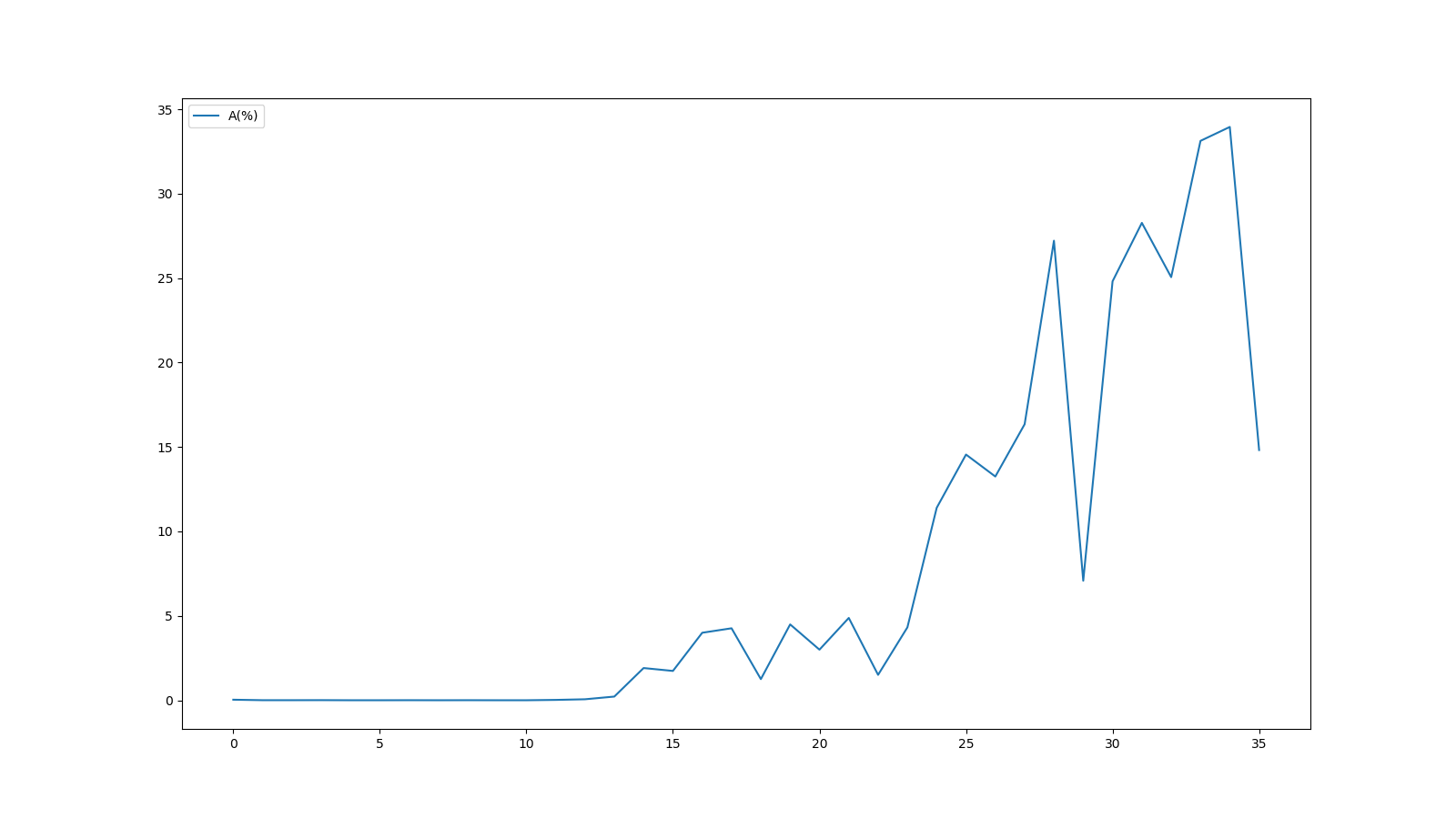
縦軸の表示範囲は絞っていますが、FastQCのグラフと同じグラフが得られたことが分かります。








コメント